Phase-locked femtosecond laser directs the outcome of a simple chemical reaction
DOI: 10.1063/1.2218534
When chemically reactive atoms or molecules combine, their valence electrons snap into new, energy-minimizing configurations. But suppose you want to steer the reaction toward a particular configuration—an unfavored oxidation state, say, or a specific isomer. Somehow, you’d have to reach into the nascent molecule and, at just the right moment, divert the valence electrons from their habitual target.
Conceivably, the intervention could be effected by means of precisely timed and targeted pulses of electric field. But to pluck and place the electrons at will, the strength, direction, and duration of the pulses should all be adjustable. On the attosecond time scale of atomic electrons, wielding such arbitrary control of electric fields is currently out of reach.
However, as a recent experiment demonstrates, one can now start approaching that ideal. The key lies in exploiting the electric field oscillations within ultrashort pulses of laser light. 1
Marc Vrakking of the FOM Institute for Atomic and Molecular Physics in Amsterdam and Ferenc Krausz of the Max Planck Institute for Quantum Optics in Garching, Germany, led the team that did the experiment. As befits a first step, the reaction is modest and the system simple: the photodissociation of the singly charged deuterium molecule D+ 2.
Ordinarily, when the D+ 2 ion breaks up, the single electron has an equal chance of ending up on either deuterium nucleus. But by adjusting the phase of the electric field within femtosecond laser pulses, Vrakking and Krausz can specify which nucleus—left or right—carries off the electron.
A more interesting question
The use of lasers to direct the movement of electrons in reacting molecules was foreshadowed by an earlier experiment. In 2002, Krausz and his collaborators used femtosecond laser pulses like a catapult to drive atomic electrons away from, then back to, the electrons’ nuclear partners. That feat yielded attosecond pulses in the extreme UV (see Physics Today, April 2003, page 27 ).
The laser that made the 2002 experiment and the current one possible emits a series of widely spaced few-femtosecond pulses, each of which contains about four oscillations. Thanks to several technological innovations, including Theodor Hänsch’s Nobel-winning frequency comb, the phase of the oscillations with respect to the amplitude envelope can be fixed.
The phase, known as the carrier envelope phase (CEP), characterizes the pattern of up-and-down oscillations. At one extreme, known as a cosine pulse, the maximum electric field and the peak of the amplitude envelope coincide. At the other extreme, known as a sine pulse, the electric field is zero at the peak of the amplitude envelope.
Although the laser’s control equipment can lock the CEP, it can’t determine the CEP’s value. To make his attosecond pulses, Krausz needed cosine pulses and used a spectroscopic method to determine the CEP. Krausz and his group then began to explore how atomic photoionziation depends on CEP.
For those experiments, Krausz positioned detectors to catch photoelectrons flying off parallel and antiparallel to the laser’s polarization axis. To detect all the photoelectrons in all directions, Krausz teamed up with Vrakking, whose ion velocity imager can do just that. As they started work, they realized their original goal—measuring the CEP dependence of ionization—suggested a more interesting question: Could the CEP control the ionization?
The experiment their team performed boils down to three steps: creating D+ 2 ions, breaking them apart in a controlled way, then recording the pattern of paths taken by D+ fragments. Deuterium was picked over hydrogen to avoid noise-producing contamination from ambient water molecules.
Krausz’s femtosecond phase-locked laser carries out the first two steps. Vrakking’s ion velocity imager, which his team installed in Krausz’s Garching lab, carries out the third step. Matthias Kling, a postdoc in Vrakking’s group, and Aart Verhoef, a graduate student in Krausz’s group, set up and ran the experiment. Figure 1 outlines how it works.

Figure 1. To demonstrate the role of carrier envelope phase (CEP) in molecular dissociation, the Amsterdam–Garching research team focused CEP-locked pulses onto a beam of deteurium molecules. The pulses have enough energy to ionize and split any molecule whose axis happens to align with the pulses’ polarization axis (the y-axis here). When the
(Adapted from ref. 1.)

Laser pulses pass first through a pair of glass wedges whose separation changes the CEP and then through a lens whose focus intercepts the beam of D2 molecules. What happens when a pulse hits a molecule depends on the molecule’s orientation with respect to the laser’s polarization. If the two axes are aligned, the laser ionizes the molecule and excites it into a state from which it splits into a D atom and a D+ ion. The ion is accelerated toward a microchannel plate detector, whose plane is parallel to the polarization. The ion’s position, along with elementary electrostatics, yields the ion’s momentum.
The right side of figure 1 shows what happens if the CEP is not locked and varies randomly from pulse to pulse. The D+ ions fly off mostly along the axis of polarization. Equal numbers go in the –y and +y directions, which means that after the D+ 2 ions dissociate, their single electrons go one way or the other with equal likelihood.
To determine whether CEP influences dissociation, Kling and Verhoef first locked the CEP, then repeated the experiment at different wedge settings, increasing the phase in steps of π/4 through 3 full cycles of 2π. To quantify the expected asymmetry of the dissociation, they integrated the counts in the 60-degree-wide segments shown in the figure. The normalized asymmetry A is defined as the difference in the segment counts divided by their sum.
Dissociation
The main panel in figure 2 maps A as a function of the kinetic energy of the D+ ions and the phase added to the CEP. Below 3 eV and above 8 eV, locking and adjusting the CEP has no influence on whether D+ ions go in the –y or +y direction. But between 3 eV and 8 eV, A oscillates between −0.2 and +0.2 with a telltale period of 2π. In that energy range, changing the CEP is like aiming the reaction left or right.

Figure 2. Whether D + ions prefer to go in the −y direction (blue), the +y direction (red), or neither direction (green), depends on the carrier envelope phase and the ions’ energy. The main panel shows that CEP-dependent asymmetry is reproducible (that is, it has a period of 2π) and that it occurs only between about 3 and 8 eV. The left panel shows that the asymmetry arises only when an electron, removed during ionization, slams back into the D2 + ion. Circularly polarized pulses, unlike linearly polarized pulses, can’t supply the D2 + with enough energy.
(Adapted from ref. 1.)
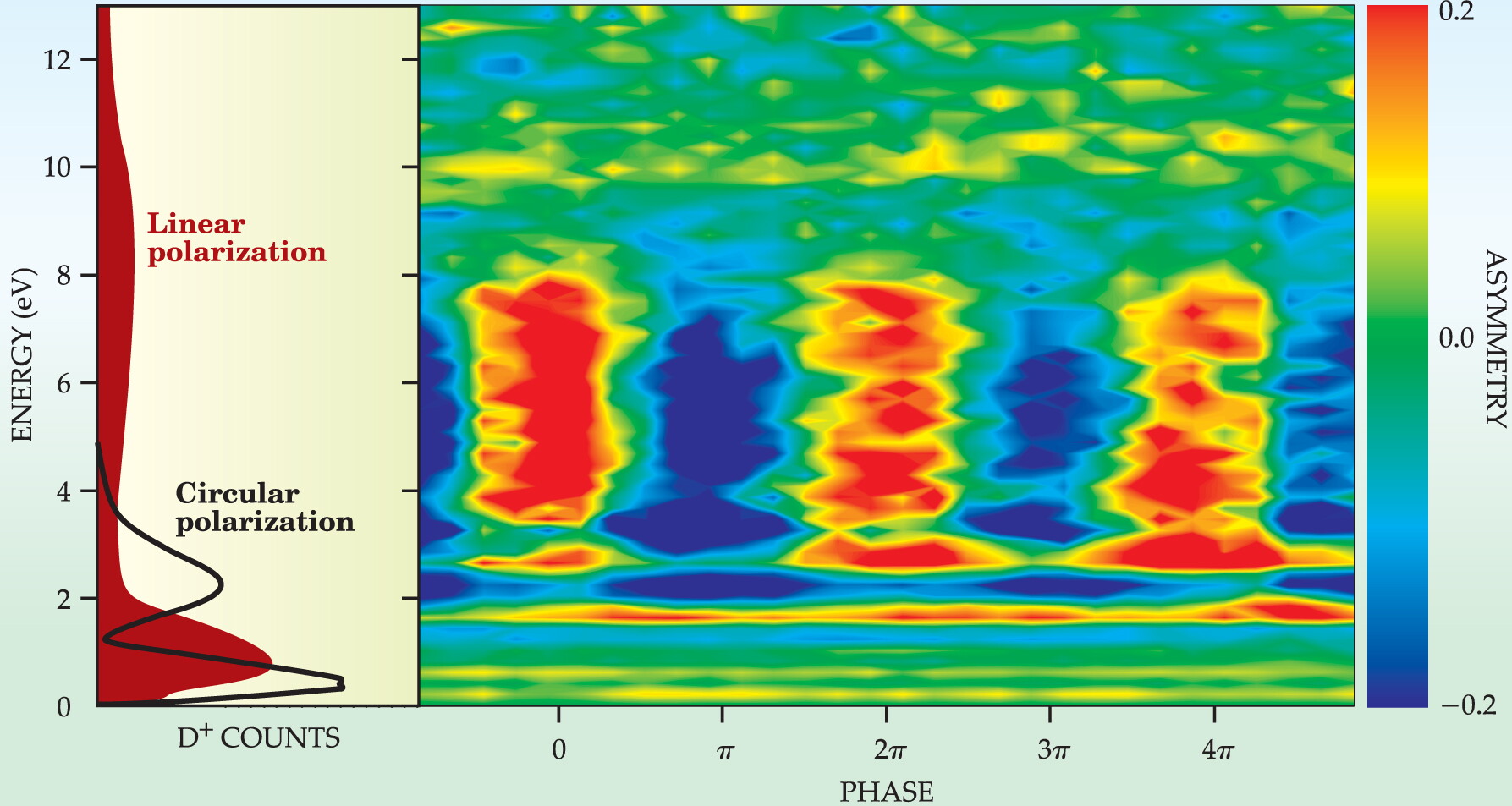
What’s so special about that energy range? The laser pulse has to ionize D2 as well as split the resulting D+ 2. At certain (and as yet unknown) values of the CEP, one of the first strong upswings of the electric field strips off an electron from the molecule. The downswing hurls the electron back at the cation, giving it extra energy that ends up, in part, boosting the D+ ion’s kinetic energy above 3 eV. Somehow the collision also makes the breakup of the cation dependent on the direction of the next peak of electric field upswing (that is, on the CEP).
The key role played by the first electron is demonstrated by the left panel in figure 2. When the laser pulse is linearly polarized, the D+ energy spectrum has a broad peak between 3 and 12 eV within which A is nonzero. But when the laser pulse is circularly polarized, the peak vanishes, presumably because a twisting field can’t hurl the first electron back at the molecule.
Vrakking’s postdoc Christian Schiedslag analyzed the breakup in terms of energy levels. Right after ionization, the D2 + sits in its
But, according to his calculations, Schiedslag believes the electric field pushed the electron partially into the 1sσ + g state, thereby breaking the wavefunction’s symmetry and introducing bias toward one D nucleus over the other. The bias depends on which way the field is pointing and hence on the CEP.
Although the recolliding electron is essential to their experiment, Vrakking and Krausz emphasize that symmetry-breaking CEP dependence can occur without the electron’s help. The field itself is what breaks the symmetry, not the first electron, whose presence is a consequence of experimental expedience: It’s easier to start with deuterium molecules than with deuterium cations.
Indeed, in a 2004 theory paper, Vladimir Roudnev, Brett Esry, and Itzik Ben-Itzhak of Kansas State University analyzed the dissociation of HD+ and H2 + and found a CEP dependence of comparable magnitude. 2 Esry would like to see Vrakking and Krausz start directly with HD+, H2 + or D2 +. Those three-body systems (two nuclei, one electron) approach the current limit for solving the time-dependent Schrödinger equation in an intense laser field.
The experimental frontier, as Krausz sees it, involves improving the ability to shape the pattern of electric field within a pulse. Broadening the frequency range down to the IR and up to the UV (at present it remains all-optical), would bring two benefits. First, at higher energies, the field itself could excite the target molecule. Second, a spectral band spanning several octaves would extend the range over which one can adjust the field’s instantaneous amplitude and frequency. The result is stronger CEP dependence and greater freedom to shape the pulse from one subfemtosecond quarter cycle to the next.
Conceivably, as Paul Haljan, Misha Ivanov, and Paul Corkum of Canada’s National Research Council proposed ten years ago, a shaped, broadband pulse could excite a molecule from its ground state to one or more excited states. 3 A later oscillation in the same pulse could then steer the excited electron inside the chemical bond, changing the atoms it would otherwise connect.
References
1. M. F. Kling et al. Science 312, 246 (2006) https://doi.org/10.1126/science.1126259 .
2. V. Roudnev, B. D. Esry, I. Ben-Itzhak, Phys. Rev. Lett. 93, 163601 (2004) https://doi.org/10.1103/PhysRevLett.93.163601 .
3. P. Haljan, M. Yu. Ivanov, P. B. Corkum, Laser Phys. 7, 1 (1997).





{kind=link}
{kind=link}