Ultrafast laser spectroscopy measures heat flow through molecular chains
DOI: 10.1063/1.2800088
Heat travels along a temperature gradient, from hot to cold, at a rate that depends on the material’s thermal conductivity. But when matter is scaled down to the level of individual atoms or molecules, the familiar concepts of heat diffusion by electrons and phonons that scatter from defects, impurities, or other excitations no longer apply. A one-dimensional molecule made up of a dozen atoms, say, carries heat through discrete vibrations over a length much shorter than the typical mean free path of electrons or phonons in a bulk molecular solid. And the vibrational modes can differ from those in the bulk material because of the reduced dimensionality and the lack of a periodic band structure. Accordingly, the thermal properties of a material at macro- and microscales can be very different.
The difference matters because long-chain molecules attached to tiny electrodes can be used to control the switching and transport of electrons in molecular-electronics devices (see the article by Jim Heath and Mark Ratner in Physics Today, May 2003, page 43 ). Localized heating from high currents can overwhelm the small heat capacity of delicate circuitry and threaten the reliability of devices if the heat isn’t dissipated efficiently.
Last year Arun Majumdar (University of California, Berkeley) and colleagues measured the heat conductance through a thin, self-assembled monolayer of long-chain hydrocarbon molecules closely packed together by weak van der Waals forces. 1 In those insulating molecules, vibrational excitations, not electrical ones, dominate the heat transport. The monolayer was sandwiched between two thermal reservoirs—gallium arsenide and gold.
Electrical resistance in a thin film varies with temperature, so Majumdar’s group could infer the thermal conductance through the organic monolayer from changes in the resistance created by an AC current applied to the gold film. The gold acted as both heater and thermometer: A value of the thermal conductance could be derived by measuring the effect of the current on the gold’s temperature.
Such a steady-state measurement, however, addresses neither the transient response of the atoms to local temperature changes along the chains nor the vibrational relaxation processes that occur. Researchers from the University of Illinois at Urbana-Champaign, led by Dana Dlott and David Cahill, have now designed an approach that captures those dynamics. Using time-resolved vibrational spectroscopy, they monitored how groups of atoms in the chains react to ultrashort and intense bursts of heat. 2
Sum-frequency generation
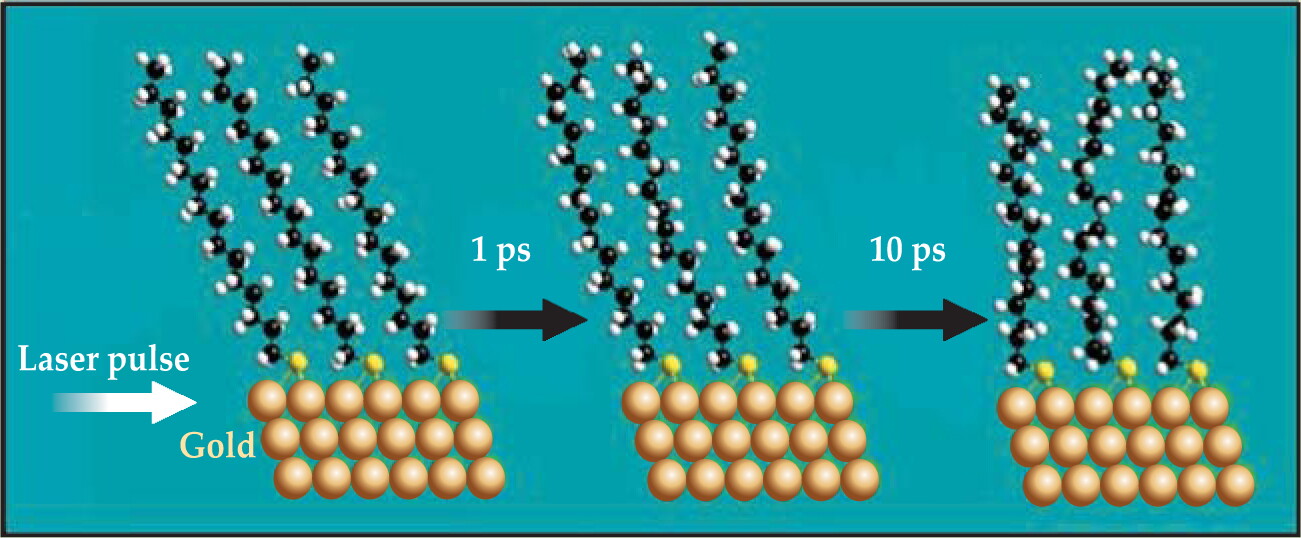
Dlott and company start, like Majumdar’s group, by growing self-assembled monolayers of S(CH2) n CH3 with varying molecular chain lengths n. But for each chain length they anchor their film to a single interface. One end of the alkane chain bonds to a gold surface through a sulfur atom, and the other end is free, terminated by a methyl (CH3) group. The methyl groups thus form a 2D surface on top of the thin film. A 500-fs laser pulse heats the gold to about 800 °C, just short of its melting temperature, as hot electrons collide inelastically and give rise to phonons in the metal. Those phonons then excite stretching and bending modes in the molecules across the interface, all on the time scale of picoseconds (see figure 1).

Figure 1. Molecular simulation of incipient heating in a layer of self-assembled alkanethiol molecules on gold. Within a picosecond of heating an initially well-ordered chain, the orientation of each methyl group starts fluctuating. The disorder in the methyl groups increases over time as atoms gradually equilibrate with each other along the chain. The picture corroborates the thermal evolution of optical spectra shown in figure
(Adapted from ref. 2.)
Using a technique known as sum-frequency generation (SFG; see also the article by Gabor Somorjai and Jeong Park on
In essence, a femtosecond IR laser pulse and a visible laser pulse intersect on the thin-film surface simultaneously, but are delayed relative to the pulse that heats the gold. When the IR pulse is tuned to the vibrational frequency of the methyl group, that part of the molecule is excited to a virtual state and emits a light signal at the sum frequency of the two pulses. In this case, the IR pulse is broad enough to cover all three vibrational transitions, from 2850 to 3000 cm−1, each of which is coherently excited. In figure 2, the transitions appear as dips against a broad nonresonant SFG background from the gold.

Figure 1. Molecular simulation of incipient heating in a layer of self-assembled alkanethiol molecules on gold. Within a picosecond of heating an initially well-ordered chain, the orientation of each methyl group starts fluctuating. The disorder in the methyl groups increases over time as atoms gradually equilibrate with each other along the chain. The picture corroborates the thermal evolution of optical spectra shown in figure
(Adapted from ref. 2.)
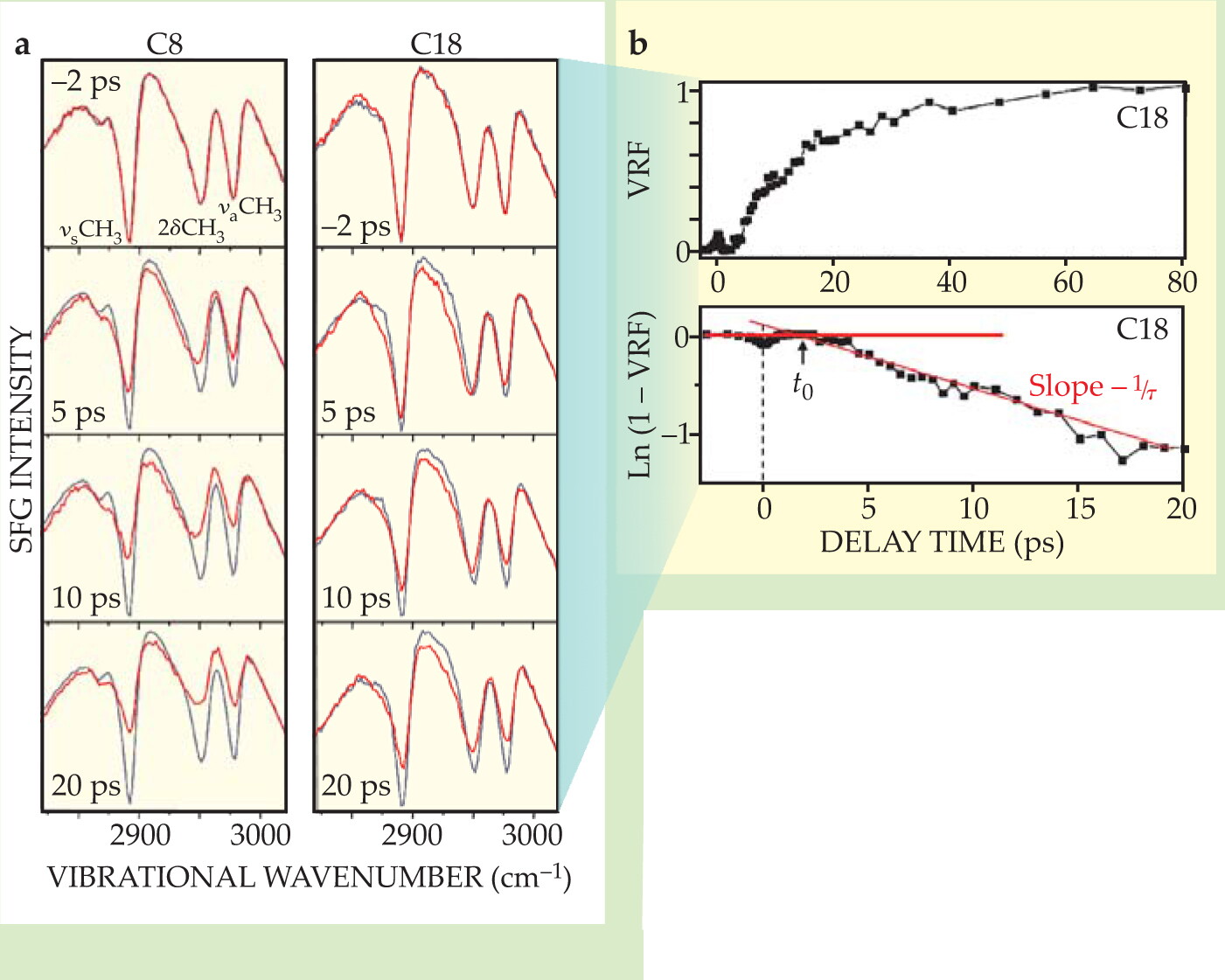
The symmetric methyl stretch in each of the roughly 1011 methyl groups in the film has an IR transition dipole moment parallel to the top C–C bond, so the observed intensity of the symmetric stretch depends on the vector sum of those dipoles—the net spatial orientation of the methyl groups. When the molecules are ordered, as they are at room temperature, the symmetric stretch appears as a deep dip in the spectrum. As heat floods into the hydrocarbon chains from the heat pulse, disorder is registered as a gradual weakening of the signal over time.
Indeed, although the vibrational states excited within a picosecond can hardly be described by an equilibrium Boltzmann distribution, Dlott argues that the orientational disorder—or rather, the ensemble-averaged IR dipole moment of the methyl groups—can be likened to an effective temperature.
The time-resolved measurement of energy flow between adsorbed molecules and surfaces goes back decades. In 1990, for instance, Philippe Guyotsionnest (now at the University of Chicago) and colleagues determined the lifetime of coupling between the silicon–hydrogen stretching mode and vibrations of the Si surface. 3 That measurement also exploited time-resolved SFG spectroscopy, but was carried out by pumping the hydrogen adsorbate vibration with a short laser pulse and recording the loss of that energy as it decayed into the surface—in effect, the inverse of Dlott’s experiment. The fundamental difference, though, lies more in the complexity of the adsorbed molecule than in the direction of energy flow. Long hydrocarbon molecules have hundreds of low- and high-frequency vibrational modes that may participate in the flow of heat.
Ballistic transport
To analyze the time evolution of the surface from the SFG data, Dlott and coworkers define a vibrational response function (VRF) and extract two characteristic times—both proportional to the chain length, it turns out. They interpret one—the time delay between the heat pulse and the first signs of disorder (t 0 in figure
The other time of interest—the time constant τ in the VRF curve—measures the time required for the vibrational excitations in chain molecules to relax and equilibrate with the gold. One can think of the gradual buildup in heat as a thermal impedance. Longer chains, with their larger heat capacities, require more time to heat up. Intriguingly, both t 0 and τ extrapolate to zero at a finite chain length of about 0.8 nm, the length of about 4 carbon bonds. Four years ago Dvira Segal, Abraham Nitzan, and Peter Hänggi predicted that the heat-carrying vibrations in short-chain alkanes should, in fact, be delocalized at about that length scale. 4
The transfer of heat through the interface, controlled by the weak coupling between gold phonons and alkane vibrations, dominates the thermal conductance. The mismatch in vibrational frequencies and density of states across the interface is probably responsible. A bulk solid’s spectrum is continuous and broadband, whereas a self-assembled monolayer’s spectrum is discrete with narrow bands. 1 And because carbon is lighter than gold, the strong C–C bonds, which transport heat along the chain, are likely to vibrate with higher frequency than metallic bonds in the gold. But as chemists and molecular engineers find innovative ways to design chains with different types of atoms and bonding configurations, the rate-limiting conductance may change from the interface to the molecule itself. That level of control would bring researchers one step closer to the goal of designing molecular chains whose thermal properties can be chemically tuned.
References
1. R. Y. Wang, R. A. Segalman, A. Majumdar, Appl. Phys. Lett. 89, 173113 (2006). https://doi.org/10.1063/1.2358856
2. Z. Wang, J. A. Carter, A. Lagutchev, Y. K. Koh, N. -H. Seong, D. G. Cahill, D. D. Dlott, Science 317, 787 (2007). https://doi.org/10.1126/science.1145220
3. P. Guyot-Sionnest, P. Dumas, Y. J. Chabal, G. S. Higashi, Phys. Rev. Lett. 64, 2156 (1990). https://doi.org/10.1103/PhysRevLett.64.2156
4. D. Segal, A. Nitzan, P. Hänggi, J. Chem. Phys. 119, 6840 (2003). https://doi.org/10.1063/1.1603211





{kind=link}
{kind=link}