Folding biomolecules are caught in the act
DOI: 10.1063/PT.3.3184
Protein folding is a central puzzle of biophysics. A linear chain of hundreds or thousands of amino acids quickly and reliably winds its way into a preordained three-dimensional shape. Yet despite decades of research, it’s not fully understood just how that happens, nor is it possible to predict a protein’s final structure from its amino-acid sequence. Solving the mystery could enable the design of artificial proteins from scratch. It could also facilitate cures for the many diseases associated with proteins misfolding into the wrong structure.
Some things are known: On the way to its final folded structure, the molecule hops among metastable, partially folded intermediates. The metastable configurations, which typically persist for tens to hundreds of milliseconds, are relatively easy to study experimentally. But the hops between them, which may harbor important clues about how proteins are guided to the correct structure, last only microseconds and are much trickier to probe. Most of what is known about those fleeting transitions comes from computational studies and indirect measurements.
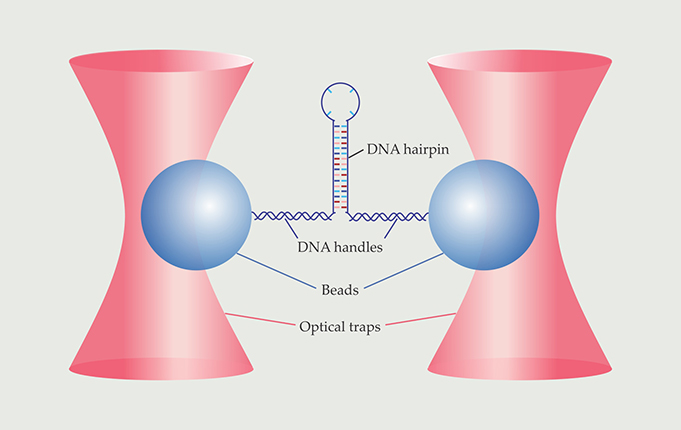
Now Michael Woodside and colleagues of the University of Alberta in Edmonton have used single-molecule force spectroscopy to observe biomolecular transition paths directly. 1 For their proof-of-principle experiment, Woodside and company used a DNA hairpin, whose structure is simpler than a protein’s, and watched it fold and unfold more than 24 000 times. As depicted in figure 1, they attached each end of a molecule to a bead held in an optical trap. The tug of the traps caused the molecule to continually flip-flop between its folded and unfolded states; from the beads’ positions at any given instant, the researchers could deduce whether the molecule was folded, unfolded, or on a transition path. Their data enabled an unprecedented statistical analysis of the time it takes the molecule to fold—a quantity that, according to Keir Neuman of the National Institutes of Health in Bethesda, Maryland, “gives access to fundamental physical aspects of the folding transition.”

Figure 1. Force-spectroscopy experiments provided the first direct look at the folding and unfolding transitions of a DNA hairpin. The hairpin was connected via two double-stranded DNA handles to beads held in stiff optical traps. The beads’ movements reflected the folding and unfolding of the hairpin. (Adapted from ref.
Foldons folding
Although protein folding in cells is sometimes assisted by chaperone molecules, most proteins can fold into their final shape without the help of any preexisting biomolecular machinery. Furthermore, proteins often partially unfold and refold spontaneously. As Cyrus Levinthal of Columbia University noted in 1969, an amino acid chain has so many degrees of freedom that finding the right folded structure through a random search of configurations would take longer than the age of the universe. There must be something about a protein’s free-energy landscape that guides it to the correct structure and makes a random search unnecessary.
The existence of distinct folding intermediates was first shown in the late 1980s through hydrogen-exchange experiments. 2 Proteins synthesized with all their hydrogen atoms replaced by deuterium were dissolved in deuterated water. While the proteins were folding, the D2O solution was diluted with H2O that swapped some of the proteins’ D atoms for H atoms. The exchange could take place on exposed, unfolded amino-acid sites, but on parts of the protein that had already folded, the D atoms were protected and stayed put. Analysis by NMR and other methods revealed the locations of the H substitutions; the experiments found that certain portions of the protein, or foldons, tended to fold all at once.
Each foldon comprises many amino acids. So in contrast to small-molecule chemistry—in which researchers speak of transition states representing the forming or breaking of a single chemical bond—the transition between protein-folding intermediates involves the rearrangement of dozens of amino-acid units and the formation of a similar number of hydrogen bonds that hold the folded structure together in its aqueous environment.
Questions still abounded: Just how long does it take a foldon to fold? Does it fold continuously or in smaller discrete steps? Is the folding process identical every time, or are there multiple distinct pathways that bridge the same two intermediates?
Fluorescence probes
Answering any of those questions requires a single-molecule experiment. Large ensembles of proteins can’t be made to go through the same folding transition at the same time, and even if they could, bulk measurements would miss crucial aspects of the transitions’ heterogeneity.
Beginning in 2009 William Eaton and colleagues at NIH have been studying protein-folding transitions using a fluorescence technique called single-molecule Förster resonance energy transfer (FRET) spectroscopy. 3 Their focus has been on small, simple proteins with only two states—folded and unfolded—with no intermediates in between. At carefully chosen points on such a protein, they attached two fluorescent dye molecules, one that emits in the green and one that emits in the red. If the green molecule is optically excited when the dyes are close enough together, it can transfer its energy to the red molecule via the FRET mechanism. The FRET efficiency, a proxy for the interdye distance, is measured by the numbers of red and green photons emitted.
The dyes are placed on the protein so that when the protein is folded, the dyes are close together and FRET efficiency is high; in the unfolded protein, the dyes are usually far apart and FRET efficiency is low. When the protein folds or unfolds, the efficiency jumps between its low and high values; the speed of the jump is a rough estimate of the duration of the transition.
Among their results, Eaton and colleagues found that the “speed of folding” in the conventional sense—how many folding transitions take place every second—has little to do with the duration of the folding transition itself. They also studied the effects of temperature and solvent viscosity to look at the role molecular friction plays in folding. But because the photophysics of the dye molecules isn’t much faster than the protein-folding transitions, the measurements offer little information about what the protein actually does between its folded and unfolded states.
Force experiments
Single-molecule force spectroscopy—which covers a whole suite of techniques for exerting a controlled tensile force on a molecule and measuring its response—has been used extensively to study the mechanical properties of biomolecules and the binding forces between them. The force can be applied and probed using optical tweezers, as in figure 1, an atomic force microscope cantilever, or various other methods.
When brought to bear on protein folding, force-spectroscopy measurements have shown that when a folded protein is pulled apart, unfolding proceeds stepwise—with each step representing an intermediate—and the distance between steps can be measured. The experiments are stable enough that researchers can watch the same molecule unfold and refold many times over the course of 20 minutes or more. But the ability to probe the transitions themselves has been hindered by the technique’s limited time resolution. In a typical optical-trap experiment, with the traps engineered to apply a constant force to the molecule, the beads take tens of microseconds to respond whenever the molecule changes length. In 2013 Matthias Rief and colleagues at the Technical University of Munich showed that the time resolution could be refined to less than 10 µs by combining stiff optical traps—whose force on the bead changes rapidly as a function of the bead’s position—with fast optical sampling and sophisticated statistical analysis. 4
For their experiments on the DNA hairpin, Woodside and colleagues used stiff traps. But because they were no longer applying a constant force, the results became more difficult to interpret—not least because the hairpin was connected to the beads by two segments of double-stranded DNA, each of which was elastic. “We had to make sure we understood well enough how the experiment worked,” says Woodside, “so we could be confident about what we were observing—especially that we were not seeing artifacts.”
To analyze their data, they first created plots of the end-to-end extension of the hairpin, as shown in figure 2a. The extension fluctuated around two separate values: the folded length xf and the unfolded length xu. The researchers derived two boundary values, x1 and x2, to mark the beginning and end of each transition.

Figure 2. As the DNA hairpin folds and unfolds, its end-to-end extension (a) hovers around two values: the folded length xf and the unfolded length xu. The boundary values x1 and x2 were chosen to represent the beginnings and ends of the transitions between the folded and unfolded states. The transitions shown in detail here (b) are representative of the more than 24 000 folding transitions and 24 000 unfolding transitions observed in total. (Adapted from ref.
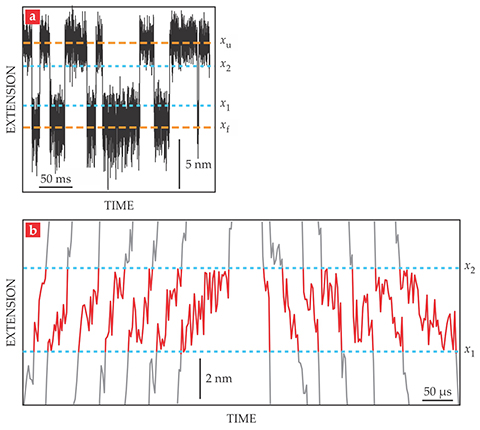
Figure 2b shows a few of the nearly 50 000 observed transitions. The average transit time, 30 µs, is the same for folding and unfolding transitions and doesn’t change when the average applied force is increased or decreased. Importantly, though, the individual transit times are not all the same. From their extensive data set, the researchers found a smooth distribution of transit times with an exponential tail reaching out to 140 µs.
An exponential distribution is expected when the folded and unfolded states are separated by an energy barrier that’s harmonic, or a concave-down parabola. Besides the duration of each transition, force spectroscopy also reveals its profile as a function of time. Some of the transitions show the molecule pausing and even doubling back on its path between the folded and unfolded states. Are those irregularities a sign of some unforeseen feature of the potential-energy landscape, or are they simply the expected result of random buffeting by water molecules? Woodside and colleagues are working on an analysis of the transition profiles to find out.
Back to proteins
As informative as the DNA hairpin experiments are, the goal is to study protein folding. So far, Woodside and company have done some force-spectroscopy measurements on the misfolding of the prion protein, the molecule associated with mad cow disease. The misfolding is a five-step process, with the unfolded and folded states separated by four intermediates; the researchers focused on just one of the steps. That step turned out to be unusually slow—with an average transit time close to a millisecond—so it was possible to operate the optical traps in constant-force mode without losing too much information from the limited time resolution. As in the case of the DNA hairpin, the distribution of transit times is consistent with a harmonic energy barrier.
Applying the method more extensively will require modifying it to suit each protein. The prion protein, like the DNA hairpin, was linked to the optically trapped beads by pieces of double-stranded DNA. But many proteins fold and unfold under such feeble forces that the DNA handles’ elasticity would obscure all the folding transitions. Alternative tethers may be necessary—as will an improved time resolution of 1 µs or finer.
Computer simulations of protein folding, which began by ignoring almost all molecular details (see Physics Today, December 2013, page 13 ), can now keep track of all atoms in a protein for appreciable lengths of time. At the same time, ever-faster time scales are becoming accessible to experiment. Woodside looks forward to when the two approaches can meet in the middle: “Direct comparisons of single-molecule experiments and simulations can not only help refine the computations but also help fill in atomistic details that are missed by the experiment.”
References
1. K. Neupane et al., Science 352, 239 (2016). https://doi.org/10.1126/science.aad0637
2. J. B. Udgaonkar, R. L. Baldwin, Nature 335, 694 (1988); https://doi.org/10.1038/335694a0
H. Roder, G. A. Elöve, S. W. Englander, Nature 335, 700 (1988). https://doi.org/10.1038/335700a03. H. S. Chung, J. M. Louis, W. A. Eaton, Proc. Natl. Acad. Sci. USA 106, 11837 (2009); https://doi.org/10.1073/pnas.0901178106
H. S. Chung et al., Science 335, 981 (2012); https://doi.org/10.1126/science.1215768
H. S. Chung, W. A. Eaton, Nature 502, 685 (2013). https://doi.org/10.1038/nature126494. G. Žoldák et al., Proc. Natl. Acad. Sci. USA 110, 18156 (2013). https://doi.org/10.1073/pnas.1311495110
More about the authors
Johanna L. Miller, jmiller@aip.org





{kind=link}
{kind=link}