Designer proteins act as logic gates
DOI: 10.1063/PT.3.4495
What if the behavior of natural and synthetic cells could be programmed like computers? Then a programmer could turn cellular behaviors on and off in a living organism by adding certain molecules. Cells already sense and respond to stimuli. But with external control, those behaviors could be put to work in medical or biotechnological tasks—for example, as smart drug delivery or telling bacteria to clean up toxic waste. To that end, researchers have engineered logic gates, largely using DNA and RNA, that introduce programmable circuitry into cells.
Although those nucleic-acid logic gates are easy to program, cells make decisions through protein–protein interactions. A protein-based logic gate can speak directly to a cell’s existing decision-making circuits. Researchers have already modified naturally occurring protein signals to introduce new logic pathways. But those implementations are inherently limited in scope by the number, properties, and geometries of the proteins.
Now David Baker of the University of Washington in Seattle and his colleagues have designed and constructed proteins from scratch that can be modularly assembled to perform an array of logic operations both inside cells,
1
as represented in figure
Figure 1.

Protein logic gates inside a living cell turn bioluminescence on and off. (Conceptual illustration courtesy of MolGraphics.)
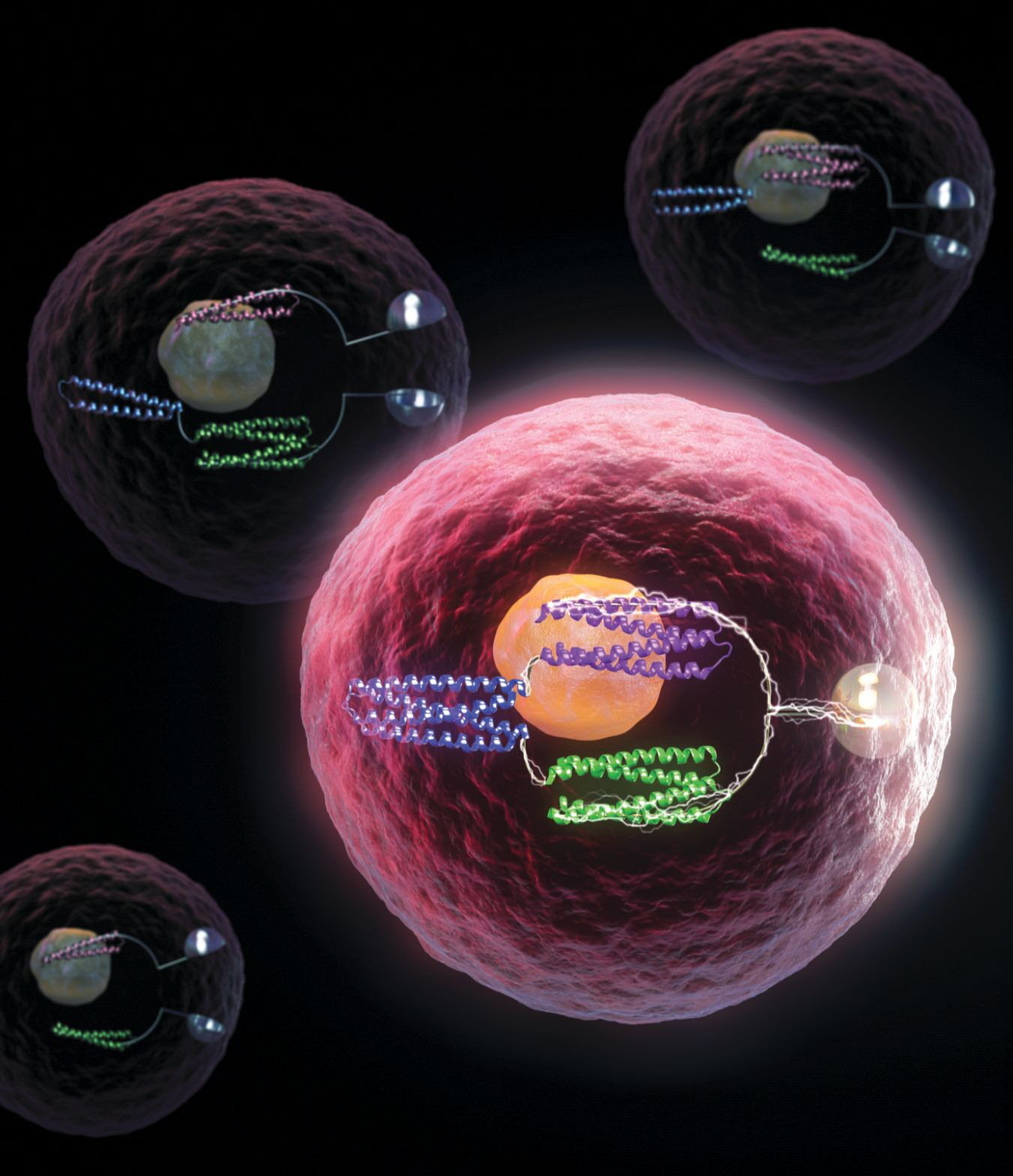
De novo design
The study’s lead author, Zibo Chen, was interested as an undergraduate in engineering protein–protein interactions, which underlie much of natural cellular decision making. As synthetic biology becomes more prevalent and complex, protein–protein interactions will need to be designed deliberately. So when he joined Baker’s lab in graduate school, Chen pursued tunable protein interactions using de novo proteins—that is, proteins designed and built from the ground up.
A protein’s particular sequence of amino acids reliably folds into the shape that minimizes energy and balances the attraction and repulsion among amino acids and the fluid surrounding them. The difficulties for researchers who design proteins from scratch are calculating the energy accurately and sorting through the large set of possible sequences: In an average-length protein, the 20 amino acids can form 20200 possible sequences, of which only 1012 occur naturally.
In the 1990s Baker and his group developed a program called Rosetta that uses a Monte Carlo sampling algorithm to solve for the lowest-energy folded structure of a given protein’s amino-acid sequence. They eventually started tackling the inverse problem—selecting a three-dimensional structure and then finding the sequence that produces it. The problem is complicated; it often takes a combination of known peptide fragments and an iteration between predicting a sequence and double-checking what structure it yields. To meet the huge computational load, Baker founded a project called Rosetta@home, in which people can offer their personal computers for protein computations. (At the moment the project is modeling SARS-CoV-2 proteins and small proteins for potential therapeutic and diagnostic uses.)
In current de novo protein design, researchers often start by picking a desired function or shape and then finding a suitable polypeptide backbone structure. The structure needs to have a high chance of being the lowest-energy state, or it won’t form in practice, and it needs to accommodate a core of amino acids. One common backbone is a bundle of helices, whose optimization is manifest in equations developed in 1953 by Francis Crick for describing how helices pack together. Once researchers select a backbone shape and solve the inverse problem for the corresponding amino-acid sequence, they perform a similar sampling process and inverse problem for the core amino acids. Finally, they manufacture or buy synthetic DNA that encodes that sequence. With that synthetic DNA, Escherichia coli bacteria produce the designed proteins.
The algorithms that design de novo proteins optimize for stability, and the resulting proteins are stable—often too stable. The protein–protein interactions necessary for logic-gate applications generally happen at interfaces that are energetically perturbed, and stable proteins resist perturbation. Protein design thus needs to strike a balance between stability and functionality.
Logical proteins
Logic gates sense and respond to inputs in a set way. To create a suite of biological logic gates, their building blocks should ideally have similar, modular structures and come in mutually orthogonal pairs. DNA is a prime example; only specific nucleotides pair together, and they all fit in a double-helical backbone. Using DNA as inspiration, Baker and his colleagues designed a collection of proteins that all took a similar form: a bundle of coiled backbones with a network of hydrogen bonds, as shown in figure
Figure 2.

Two designer protein monomers A and A′ (green and purple coils) form a dimer through a network of complementary hydrogen bonds, as shown in the inset. The heterodimer they form, A:A′, is represented by the interlocked symbols shown in blue. (Adapted from ref.
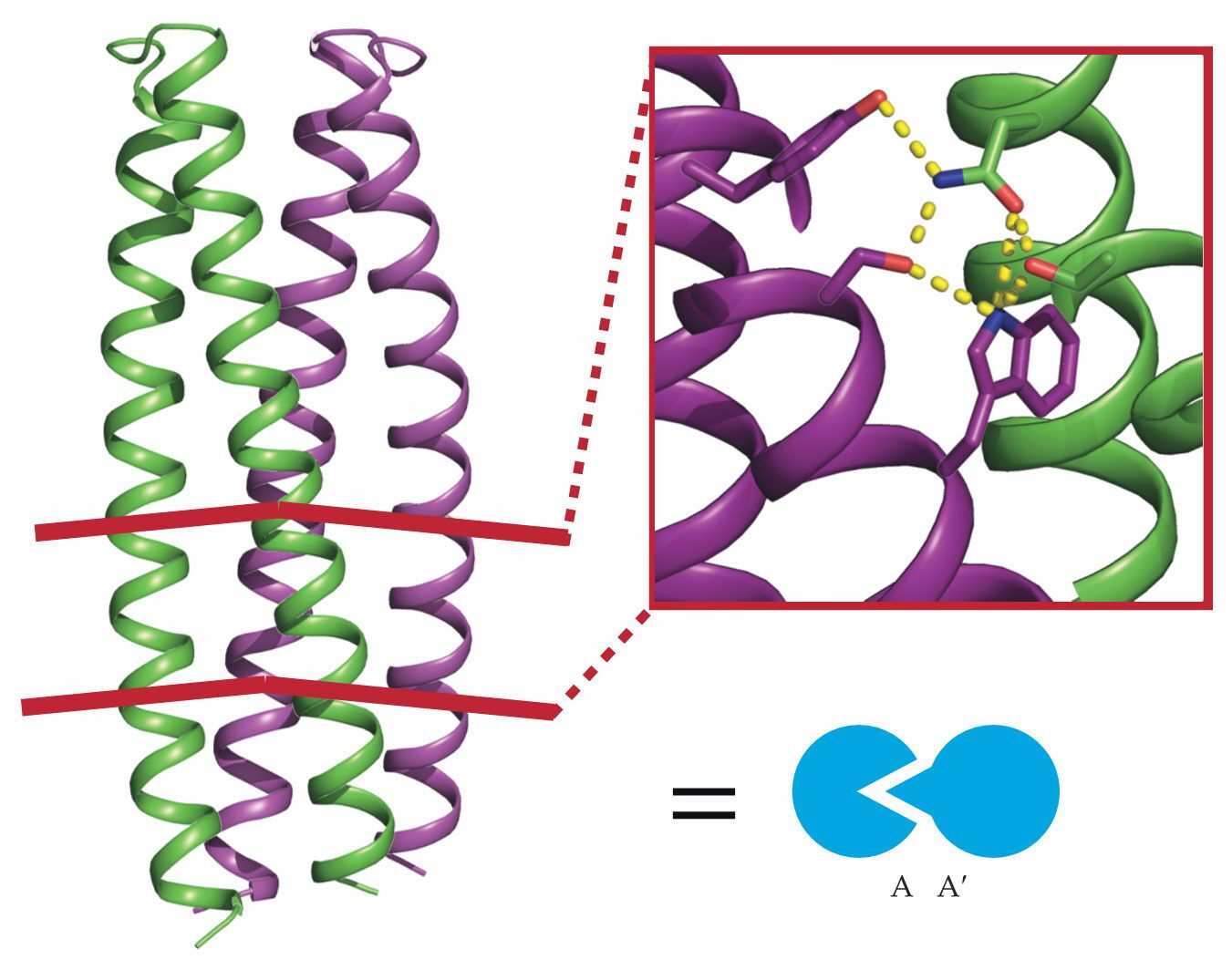
In 2016 Scott Boyken, then Baker’s postdoc, introduced a computational method to enumerate all the possible hydrogen bond networks for a given backbone structure. Boyken, Chen, and their colleagues then employed the technique to design a set of 39 orthogonal protein pairs. 2 With more pairs than, say, DNA’s two pairs of nucleotides, gate complexity has fewer limits. Because the networks were designed with atomic-level accuracy, far more than those 39 pairs are possible.
Turning protein pairings into logic gates takes judicious combinations of complementary monomers. For example, take three pairs of monomers that form the heterodimers A:A′, B:B′, and C:C′. With complexes of A′ and C monomers (A′–C) and C′ and B′ monomers (C′–B′) as inputs, the system acts as an AND gate, as shown in figure
Figure 3.

AND and OR gates can be constructed from combinations of isolated and linked protein monomers. (a) An AND gate can be made from three pairs of complementary monomers—A:A′, B:B′, and C:C′. In the presence of A and B monomers, complexes of A′ and C monomers (A′–C) and C′ and B′ monomers (C′–B′) serve as inputs. A and B are joined, an output signal of 1, only if all monomers form a single complex, as in the bottom row. (b) Surrounded by E′ monomers and A–D complexes, the inputs A′–E and D′–E produce a single output complex —a signal of 1—when at least one input is present, as in an OR gate. (Adapted from ref.
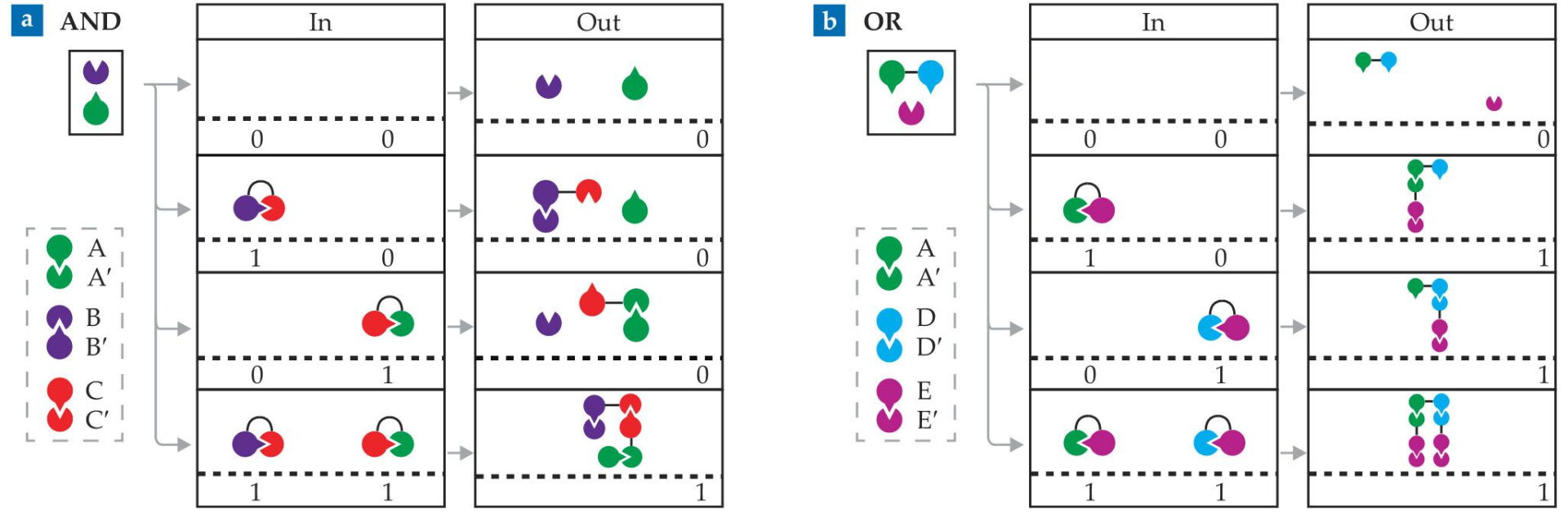
The signal from those fully bonded complexes depends on what researchers bind to the noninput monomers. For example, Baker and his colleagues constructed an AND gate in a yeast cell containing a transcription protein with two separable parts. They fused one part to the A monomer and the other part to the B monomer. In the positive output configuration, those two parts are close together, and that positioning activates a gene responsible for increasing the yeast’s growth rate. Otherwise, the growth rate stays the same.
In similar fashion to the AND gate, an OR gate can be devised, as shown in figure
Cellular output
To work in a cell, the logic gate can’t be too sensitive to population imbalances in the available monomers. So monomers must be tied in a suitable way, such as with an optimized length of linker and with the binding interfaces hidden. For two properly fused monomers, the energy required to expose their bonding sites is provided by the sum of the binding energies for their complementary monomers—for example, the A′–E complex unfolds only if both A and E′ are present; if only A is present, the energy barrier is too high, and A′–E won’t unfold. Partial complexes won’t form, and a major imbalance in the inputs won’t throw off the gate’s function.
To test that cooperativity in actual samples, the researchers used native mass spectrometry, which measures the populations of different compounds in their nearly native state. They found that even a sixfold imbalance in the inputs didn’t affect the number of fully bonded complexes.
Biological logic gates could be important for medical treatments. As a demonstration, the researchers used human T cells, essential to immune responses. When T cells fight chronic infections and cancer, they often suffer from a dysfunction known as exhaustion, in which there’s a sustained inhibitory signal that stops T cells from doing their job. 3 But T cells need transient inhibitory signals to prevent autoimmune disorders. By modulating their logic-gate inputs, Baker and his group were able to selectively repress a gene thought to modulate exhaustion. With their logic gate, T cells may be able to overcome inhibitory signals only in the case of exhaustion.
References
1. Z. Chen et al., Science 368, 78 (2020). https://doi.org/10.1126/science.aay2790
2. Z. Chen et al., Nature 565, 106 (2019). https://doi.org/10.1038/s41586-018-0802-y
3. E. J. Wherry, M. Kurachi, Nat. Rev. Immunol. 15, 486 (2015). https://doi.org/10.1038/nri3862





{kind=link}
{kind=link}
{kind=link}