Deep learning opens up protein science’s next frontiers
DOI: 10.1063/PT.3.4850
At their heart, proteins are much like any other polymers: flexible linear chains of amino-acid monomers drawn from a library of just 20 or so building blocks. But unlike synthetic polymers, which tend to flop around stochastically, proteins reliably fold into characteristic three-dimensional shapes. The diversity of those shapes gives rise to the complexity of the biological world.
Uncovering the relationship between amino-acid sequence and folded structure has been a grand challenge of the past half century, with connections to cell biology, chemistry, biophysics, and medicine. To date, more than 180 000 protein structures have been made available to the world in the Protein Data Bank (PDB). But even that enormous resource barely makes a dent in the tens of millions of proteins known to be encoded by genes across all living species.
Last November, as part of the Critical Assessment of Structure Prediction (CASP) project, researchers at DeepMind in London showed that their AlphaFold2 model had made astonishing progress. Given a protein’s amino-acid sequence, AlphaFold2 could often predict its structure with most atomic positions correct to within an angstrom—less than the length of a chemical bond.
1
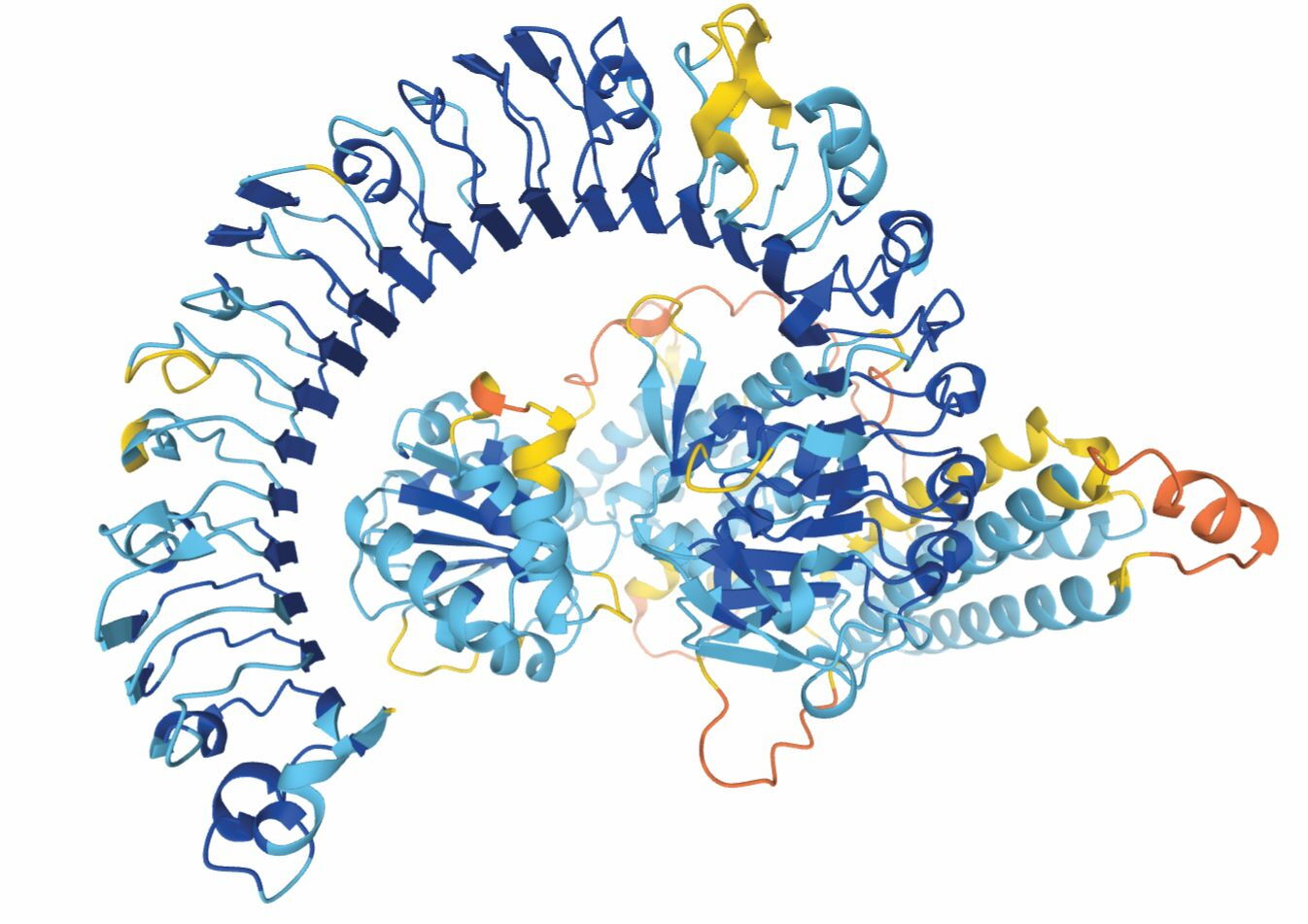
The team has now released its own database of predicted protein structures, including the complete human proteome and many nonhuman proteins whose structures, such as the one in figure
Figure 1.

From AlphaFold’s vast collection of protein structure predictions comes this protein, which may promote disease resistance in the Eurasian wildflower Arabidopsis thaliana. The colors represent regions of the protein predicted with high (blue) through low (yellow and orange) confidence. The structure has yet to be observed experimentally. (Courtesy of the AlphaFold Protein Structure Database, CC BY 4.0 .)
Inspired by AlphaFold2 but with only a rough idea of the model’s architecture, Minkyung Baek, in David Baker’s group at the University of Washington, and her colleagues developed a similarly capable model, called RoseTTAFold, in time to publish their results concurrently with AlphaFold2’s this summer. 2
Both AlphaFold2 and RoseTTAFold use deep learning—a type of artificial intelligence—which means that their inner workings are largely a black box. But their guiding principles are some of the same ones that have been guiding structural biologists for years. And their success has researchers thinking about how to paint an even more complete picture of proteins and their biological environments.
A hard problem
Compared with the rest of organic chemistry, understanding of proteins came late. The first known protein structures, of myoglobin and hemoglobin, weren’t discovered until 1958 and 1959, respectively—half a decade after the structure of DNA.
And unlike DNA’s elegant double helix, protein structures were a mess. Linus Pauling, among others, had predicted years earlier that amino-acid chains could organize into orderly alpha helices and beta sheets. Indeed, those motifs do show up in protein structures, but they’re interspersed with wild twists and turns that hadn’t been anticipated. “There was a sense of, ‘Holy cow, there’s no symmetries here,’” says Ken Dill of Stony Brook University. But the structure wasn’t disordered either: For any given protein, it was always the same.
The hemoglobin and myoglobin structures had been found through x-ray crystallography, the long-time gold standard for probing the atomic structure of any material, not just biomolecules. (See the article by Wayne Hendrickson, Physics Today, November 1995, page 42 .) But the powerful technique is beleaguered by a pair of challenges. First, it requires a crystalline sample—an unnatural form of matter for most proteins. Second, the x-ray diffraction pattern retains only half the crystal’s structural information: The x rays’ intensities are easily measured, but their phases are lost. Max Perutz, discoverer of the hemoglobin structure, solved the so-called phase problem by inserting various heavy-metal atoms into the protein to scramble the phases. (See Perutz’s obituary in Physics Today, August 2002, page 62. ) But that trick doesn’t always work.
In recent years, cryoelectron microscopy has started to rival x-ray crystallography in its ability to image proteins with atomic resolution. (See Physics Today, December 2017, page 22 .) It has the benefit of not needing a crystal—instead, the molecules are embedded in a thin sheet of vitreous ice—but it’s still challenging. The folded proteins might unravel under the effects of surface tension, and one needs to computationally align many 2D images at random angles to convert them into a composite 3D structure. Finding protein structures experimentally by any method remains difficult and laborious.
What about theoretically? Although some proteins require chaperone molecules to fold correctly, most can solve their own folding problem using nothing but the laws of physics. It’s tempting to try to reproduce their solutions in a computer simulation (see Physics Today, December 2013, page 13 ), but the complexity of the system quickly runs up against the limits of computer power for all but the smallest proteins.
And the physics of folding is subtle. The folded structure is generally the thermodynamically favored one, but not by much, and the details of interatomic forces are extremely important. “If you don’t know the forces, then a bigger computer doesn’t help you get the right answers,” says Dill. “It just gives the wrong answers a lot faster.”
Biological shortcuts
Any successful approach to protein-structure prediction, including the new deep-learning models, needs to draw on insights from biology, not just chemistry and physics. “Protein sequences aren’t random. They’ve been distilled by natural selection,” says Temple University’s Vincenzo Carnevale. Any given protein has thousands of evolutionary cousins from across the tree of life; that evolutionary context can provide hints about structure.
Proteins with similar sequences probably have similar structures. If the structure of a related, homologous protein is already known, it can be used as a template: The new sequence is fitted into the old structure, then adjusted accordingly. “That works in a stupendous way,” says Carnevale, “but only because the community realized that to succeed with this approach, we needed not just to increase the size of the PDB but to explore the right regions of protein-sequence space.”
From 2000 until 2015, the Protein Structure Initiative guided the discovery of thousands of new protein structures, chosen not haphazardly but with the goal of systematically exploring the possible structures proteins can adopt. By the end of the project, says Carnevale, “there was no longer anything novel being discovered in protein structural space. It had all been exhaustively mapped out.” Although not every protein sequence has a sufficiently similar known structure to use as a template, the days of wholly unanticipated new protein structures were past.
Meanwhile, theorists were working on ways to glean information about a protein structure from its evolutionary context even when none of its relatives’ structures are known. That surprising feat is possible because each protein in a family is the product of its own evolutionary optimization. When one amino acid in a protein randomly mutates, the mutation usually isn’t enough to ruin the entire structure, but it does destabilize it. Evolutionary pressure therefore builds on the amino acid’s neighbors in 3D space to mutate also and thus restore the structure’s stability. If, in a list of many related protein sequences, two amino-acid positions show a tendency to mutate in tandem, they likely sit next to each other in the folded protein.
Structural revolution
Those methods and others were already being tried and tested at CASP before DeepMind entered the fray. Part computational experiment, part competition, CASP challenges hundreds of research groups every two years to reproduce protein structures that have recently been found experimentally but not yet published. Target structures are classified by difficulty, based in part on whether a homologous structure exists to use as a template. Structure predictions are graded on a scale from 0 to 100: A random guess might score below 20; an atomically precise structure, above 90.
From the early days of CASP, models have been scoring above 80 for the easiest template-based predictions, while scores for the most difficult targets have been stuck around 40. So DeepMind’s first CASP entry, the original AlphaFold model in 2018, shook things up by scoring above 70 for more than half of the most difficult targets. 3
The power of deep learning is that it can recognize patterns, interpolate between known structures, and identify mutation correlations more keenly than human observers or more straightforward algorithms can. AlphaFold wasn’t the first machine-learning model to be entered into CASP; its superior performance came, in part, from using the structure and correlation data to predict not just which pairs of amino acids are in contact but the full matrix of all their pairwise distances.
For the 2020 CASP assessment, the DeepMind team had revamped its model into AlphaFold2, whose predictions scored near 90 even for the most difficult targets—scores so high that they were probably limited by the imprecision of the experimental structures the predictions were graded against.
The AlphaFold2 code and method weren’t made public at first; all that was released to the world was a 30-minute presentation that described a model that processed data on two parallel tracks. One carried the list of protein sequences thought to be related to the target protein; the other, a pairwise amino-acid distance matrix. By exchanging information between the two tracks, the model repeatedly updated both sets of data until it converged on a final prediction, from which a 3D structure was extracted.
Building on the ideas in the presentation, Baek and colleagues developed RoseTTAFold: a three-track model that iteratively updates the sequence data, distance matrix, and 3D structure itself. If it had been entered into CASP in 2020, its scores for the hardest targets would have averaged about 80.
Now that the AlphaFold2 details are published, Baek concedes that it’s a better engineered method. “Almost every component is based on some physical insight,” she says. For example, AlphaFold2 requires its amino-acid distances to satisfy the triangle inequality—two points can’t be farther from each other than the sum of their distances to a third point—so it saves time by maintaining a degree of physicality even at intermediate steps.
Furthermore, while RoseTTAFold was trained on all the PDB structures, AlphaFold2’s training data included additional structures predicted by the model itself. “Training data is very critical,” says Baek, “so I think that more complete coverage of protein space helped them a lot.”
Molecular interactions
Has deep learning solved the notorious protein-folding problem? That depends on how the problem is defined. Dill draws a distinction between predicting protein structures—what AlphaFold2 and RoseTTAFold do—and understanding protein folding. He considers the latter, which involves mapping the funnel-shaped energy landscapes that guide amino-acid chains into their folded structures, to be largely solved already by statistical physics. 4
As far as structure prediction is concerned, the deep-learning models have reached a milestone, but they’re far from the finish line. Proteins in nature aren’t isolated structures. They interact with surrounding molecules, including water, and they combine with other proteins to build large molecular machines—and, ultimately, multicellular living organisms.
Deep-learning methods have made some headway toward solving the structures of multimolecular complexes: The structure in figure
Figure 2.

RoseTTAFold generated this structure of the human signaling protein interleukin-12 (purple) bound to its receptor (blue). Although the structures of molecular complexes are tougher to predict than those of single proteins, the structure here agrees well with one found experimentally through cryoelectron microscopy. (Courtesy of Ian Haydon, Institute for Protein Design, University of Washington.)

“Experimental methods are by no means obsolete,” says DeepMind scientist Kathryn Tunyasuvunakool. “They can provide information that AlphaFold currently can’t.” The model’s big advantage, she says, is that it produces structural starting points quickly—in minutes, rather than months or years—and in large numbers. “That’s useful, for example, for generating hypotheses and planning experiments.” The deep-learning models are already helping experimenters to fill in the missing structural data from their x-ray crystallography and cryoelectron microscopy experiments and to tackle ever more challenging structure problems.
New drugs
One of the most important uses of protein structures is in drug development. To stop a protein from performing some harmful action in the body, pharmaceutical scientists study the protein’s structure, identify a nook or cranny that may correspond to the protein’s active site, and design a molecule to plug it up like a cork in a wine bottle.
“But there’s really no such thing as ‘the’ structure,” says Carnevale, because proteins flex and contort. Focusing on fitting a molecule to one static structure ignores all the other conformations a protein can adopt or the transitions between them, any one of which might offer a more effective way to disrupt the protein’s function.
In some cases, the dynamic approach to drug development might be the only viable one. In neurodegenerative conditions like Alzheimer’s and Parkinson’s diseases, amino-acid chains get tangled up and fold into the wrong structure, called an amyloid fibril. The fibril structure is known (see Physics Today, June 2013, page 16 ), but the structure alone doesn’t say much about how the fibril forms—or how to stop it from forming.
It would take a far more sophisticated model than the ones available today to predict a protein’s entire conformational ensemble and range of motion. But as Carnevale points out, “Surely the sequence must encode that information, because nature knows what it is.”
Another ambitious goal that’s on Baek and colleagues’ minds is to free their model from the need to consider evolutionary relationships at all and predict the folded structure based only on the amino-acid sequence. Evolution has produced a wondrous array of proteins and functions, but it hasn’t come close to exploring every possible protein. The Baker lab’s specialty is in designing proteins from scratch to do things that natural ones can’t. (See Physics Today, June 2020, page 17 .) But those bespoke proteins don’t come with millions of years of evolutionary relatives to analyze.
Says Dill, “The whole field is headed toward bigger, better, faster”: bigger proteins, more complex actions, and more detailed information than has ever been possible before. Lately, modelers and experimenters alike have been working on understanding the spike protein of SARS-CoV-2—the virus that causes COVID-19—whose binding to a host cell involves a cascade of large conformational changes. 5 As Dill explains, “It’s a huge protein that’s part of an even huger complex, the virus, with all kinds of moving parts like a big Rube Goldberg machine.”
References
1. J. Jumper et al., Nature 596, 583 (2021); https://doi.org/10.1038/s41586-021-03819-2
K. Tunyasuvunakool et al., Nature 596, 590 (2021). https://doi.org/10.1038/s41586-021-03828-12. M. Baek et al., Science 373, 871 (2021). https://doi.org/10.1126/science.abj8754
3. A. W. Senior et al., Nature 577, 706 (2020). https://doi.org/10.1038/s41586-019-1923-7
4. R. Nassar et al., J. Mol. Biol., 167126 (2021), doi:https://doi.org/10.1016/j.jmb.2021.167126 .
5. See, for example, E. Brini, C. Simmerling, K. Dill, Science 370, eaaz3041 (2020); https://doi.org/10.1126/science.aaz3041
T. Sztain et al., Nat. Chem. (2021), doi:https://doi.org/10.1038/s41557-021-00758-3 .
More about the authors
Johanna L. Miller, jmiller@aip.org





{kind=link}
{kind=link}