Chemistry Nobel honors protein design and modeling
DOI: 10.1063/pt.gxte.zzen

David Baker, Demis Hassabis, and John Jumper are the recipients of the 2024 Nobel Prize in Chemistry for their work on protein design and structure. Baker, of the University of Washington in Seattle, receives half the prize for using computational methods to design proteins. Hassabis and Jumper, at Google DeepMind in London, share the other half for predicting protein structures with neural network–based AI, the method that earned its developers this year’s Nobel Prize in Physics (see page 12 of this issue).
David Baker IAN C. HAYDON/UW MEDICINE Demis Hassabis GOOGLE DEEPMIND John Jumper GOOGLE DEEPMIND





To design amino acid sequences that would fold into desired structures, the earliest computational approaches searched through a vast number of sequences for a few that, when combined together, could yield new 3D structures. To make the process more efficient, Baker and his research group developed the Rosetta computer program in the 1990s. It takes structural-fragment data from various existing proteins and uses an energy-optimization procedure to assemble them together into a new form. 1 , 2 With Rosetta, Baker and colleagues created an entirely new and large protein known as Top7: Its sequencing and structure were significantly different from the proteins found in the research community’s databases.
When Rosetta was developed, computational resources were meager enough that Baker founded a citizen science project called Rosetta@home. It’s still in use today, and people can donate computational power from their personal computers to help complete Rosetta’s calculations. Before the work by Baker’s team, protein engineers had relied mostly on the modification of naturally occurring proteins—the achievement behind the 2018 chemistry Nobel (see Physics Today, December 2018, page 22 ). The de novo design capabilities ushered in by Rosetta have allowed biochemists to build proteins from scratch. 3 Such engineered proteins can be used for various functions, such as performing logic operations inside human cells (see Physics Today, June 2020, page 17 ).
So much of a protein’s function depends on its 3D structure. Starting in the 1950s, researchers have used methods such as x-ray crystallography to empirically identify protein structure. But even today, x-ray crystallography is time-consuming, and not all proteins can be crystallized for the measurements. Since 1994, protein researchers have held a biennial challenge—the Critical Assessment of Structure Prediction (CASP) experiment—to improve theoretical structure predictions by testing them against experimental observations. The prediction accuracy, graded on a scale of 0 to 100, has been steadily improving for years, in part because AI models are adept at pattern recognition.

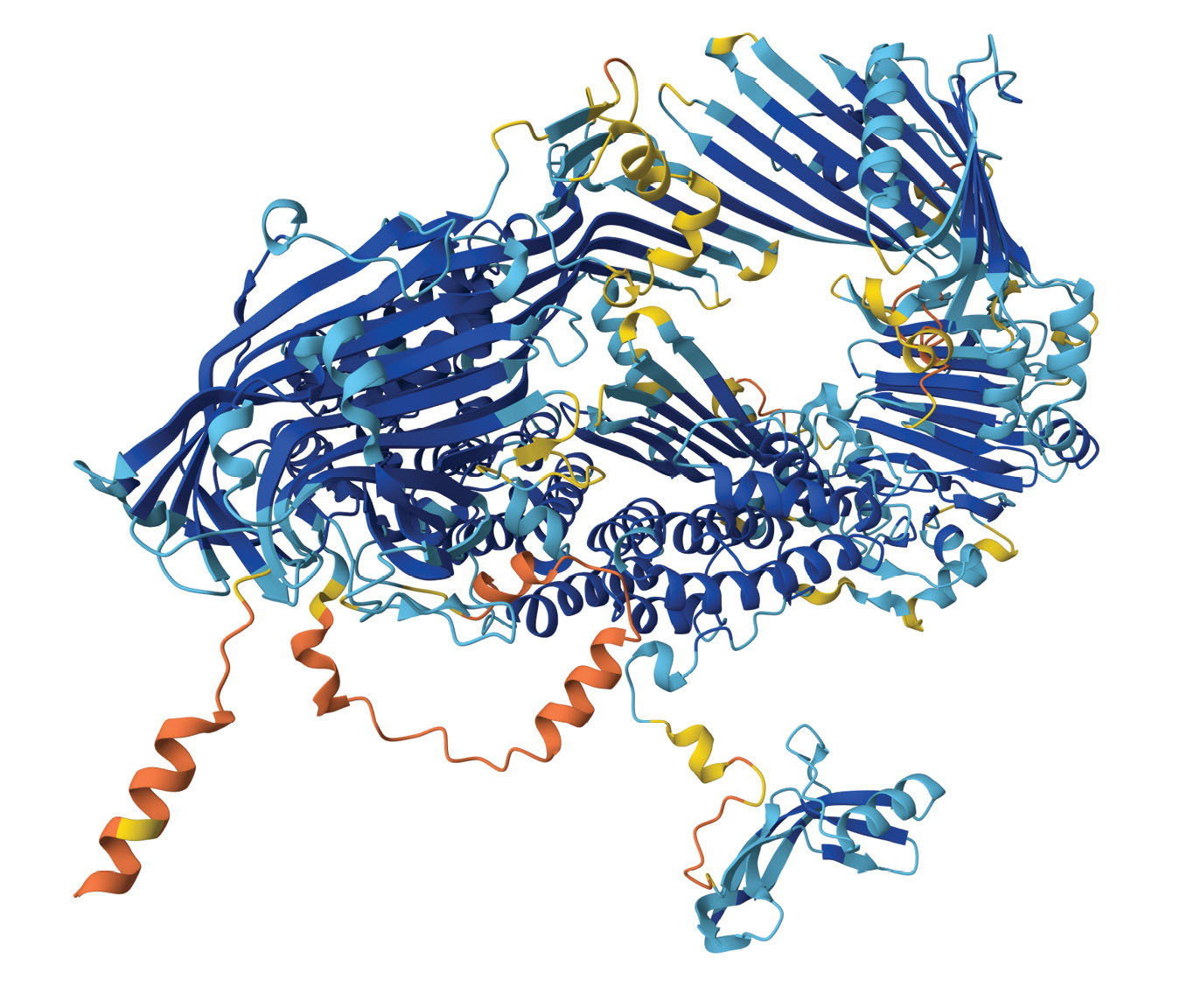
AlphaFold2’s predicted structure of the vitellogenin protein, which contributes to immunity in honeybees. The colors represent regions of the protein predicted with high (blue) through low (yellow and orange) confidence. (AlphaFold Protein Structure Database/CC BY 4.0 .)
From 2016 to 2020 at DeepMind, an interdisciplinary team led by Hassabis and Jumper reworked its neural network algorithm to dramatically improve the prediction of protein structures. 4 , 5 The transformative work was published in 2021—unusually recent for a Nobel Prize. 6 For the 2018 CASP experiment, DeepMind’s original AlphaFold model predicted protein targets better than any other model, receiving a grade of almost 70 for the most difficult targets. (In the previous CASP experiment, the best model in that category scored around 40.) DeepMind overhauled the AI algorithm for the 2020 CASP assessment, and AlphaFold2 leaped ahead of the competition and for the most difficult targets scored near 90—that’s a grade equivalent to that of an experimentally determined structure. The prizewinning research was covered in detail in Physics Today in October 2021 (page 14) .
The accurate design of proteins and the prediction of their structure have far-reaching implications in medicine and many other fields—the vitellogenin protein shown above, for example, is important for the immunity of honeybees. Earlier this year, Hassabis, Jumper, and colleagues reported the development of AlphaFold3, an upgraded model that also predicts the structures of biochemical complexes that contain nucleic acids, small molecules, and other components. 7
Hassabis has a background in computer science and video game design. Before studying proteins and AI, Jumper majored in physics and mathematics. Only after dropping out of a physics PhD program did he become interested in biology. “I think of myself as a physicist who likes to work on these really complex, really interesting systems of biology,” says Jumper. When asked what led him to join DeepMind, he recalls thinking that “machine learning and AI were exciting, and they were possibly going to solve some really important problems, and DeepMind was the most exciting place to do it.”
References
1. K. T. Simons et al., Proteins: Struct. Funct. Bioinform. Suppl. 37, 171 (1999). https://doi.org/10.1002/(SICI)1097-0134(1999)37:3+<171::AID-PROT21>3.0.CO;2-Z
2. B. Kuhlman et al., Science 302, 1364 (2003). https://doi.org/10.1126/science.1089427
3. P.-S. Huang, S. E. Boyken, D. Baker, Nature 537, 320 (2016). https://doi.org/10.1038/nature19946
4. A. W. Senior et al., Nature 577, 706 (2020). https://doi.org/10.1038/s41586-019-1923-7
5. J. Jumper et al., Nature 596, 583 (2021). https://doi.org/10.1038/s41586-021-03819-2
6. L. Tozer, “Scientists are waiting longer than ever to receive a Nobel,” Nature, 29 September 2023. https://doi.org/10.1038/d41586-023-03086-3
7. J. Abramson et al., Nature 630, 493 (2024). https://doi.org/10.1038/s41586-024-07487-w
More about the authors
Alex Lopatka, alopatka@aip.org





{kind=link}
{kind=link}
{kind=link}
{kind=link}