Spectroscopy shines light on an electrode–water interface
DOI: 10.1063/PT.3.4936

The energy available in 1 kg of molecular hydrogen gas is the same as in almost 3 kg of gasoline. Hydrogen’s superior energy density has attracted the attention of scientists and engineers working to develop more efficient and sustainable energy systems to replace fossil fuels (see the article by Joan Ogden, Physics Today, April 2002, page 69 ).
One way to boost H2 production is electrocatalysis, which drives chemical reactions with an electric potential at a liquid–solid interface. In the hydrogen-evolution reaction, two protons and two electrons combine at a cathode to yield H2. The reaction rate strongly depends on interfacial water—that is, the water in the immediate vicinity of a solid electrode’s surface. It behaves differently from the bulk and forms a layered, ordered structure.
Chemists have used various spectroscopic methods over the years to try to better understand how the specific structure interacts with cations in the electrolyte to regulate the hydrogen-evolution reaction. But many of those methods are limited to working at a modest range of electric potentials near the value for which an electrode lacks any free charge at its surface. Those conditions are quite different from the electric overpotentials that interfacial water experiences on single-crystal surfaces in cells designed for producing H2.
Now Feng Pan of Peking University in Shenzhen, Jian-Feng Li of Xiamen University, both in China, and their colleagues have measured interfacial water at conditions more suitable for producing H2. When sodium ions are added to the solution, their interference with hydrogen bonding helps form the water’s structure. 1 Those findings may affect the electrolyte chosen to promote water dissociation, lead to enhancements in the rate and efficiency of the hydrogen-evolution reaction, and further improve hydrogen-fuel production.
SHINERS
Surface-enhanced Raman spectroscopy (SERS) has been the standard method to characterize interfacial water. When light shines on a metal with water or other molecules adsorbed to the surface, conduction electrons at the interface are stimulated by the incident light and generate a surface plasmon resonance. Electromagnetic theory predicts that the plasmon resonance enhances Raman scattering at a surface. Chemical theorists, on the other hand, have argued that the enhancement arises from a transfer of charge at the surface. (See the article by Katrin Kneipp, Physics Today, November 2007, page 40 .)
Regardless of the exact mechanism, the practical result is that the light interaction excites a molecule into a higher-energy vibrational state. That signal can be exploited to identify the constituent atoms, bond lengths, and molecular structures.
With SERS, even single molecules are detectable. But the typical atomically flat single crystals that are used as reaction surfaces for studying the hydrogen-evolution reaction cannot host the necessary surface plasmon resonance that SERS requires. And other spectroscopy techniques are only effective at measuring interfacial water at relatively low electric potentials.
To collect their data, Pan, Li, and their colleagues turned to a Raman-spectroscopy technique that Li helped design in 2010. That method, shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS), uses a layer of gold nanoparticles to amplify the Raman scattering signal. 2 To prevent any unwanted chemical interactions between the interfacial water and the gold surfaces, the researchers coated each nanoparticle with a 2 nm shell of inert silica. Li says, “The SHINERS technique overcomes the long-standing limitations of SERS and can precisely characterize various materials, especially those on single-crystal surfaces.”
The researchers focused on measuring interfacial water’s structure by preparing a 50-µm-thick electrolyte solution, which shares little to no behavior with bulk water, and trapping it between two electrode surfaces. The formation of H2 bubbles interfered with previous spectroscopy measurements, but the vertically oriented reaction cell the researchers designed mitigated that problem.
Figure
Figure 1.

A sodium ion (blue) helps split a nearby water molecule, a process that accelerates the formation of molecular hydrogen at a water–electrode interface. At strongly negative electric potentials and high sodium concentrations near a palladium–gold electrode surface (silver and orange circles, respectively), sodium cations help pull water molecules toward the interface. Once there, adsorbed hydrogen strongly bonds to the palladium. That bond promotes water dissociation and, through a series of steps, the production of H2 gas. (Adapted from M. M. Waegele, Nature 600, 43, 2021, doi:10.1038/d41586-021-03511-5 .)
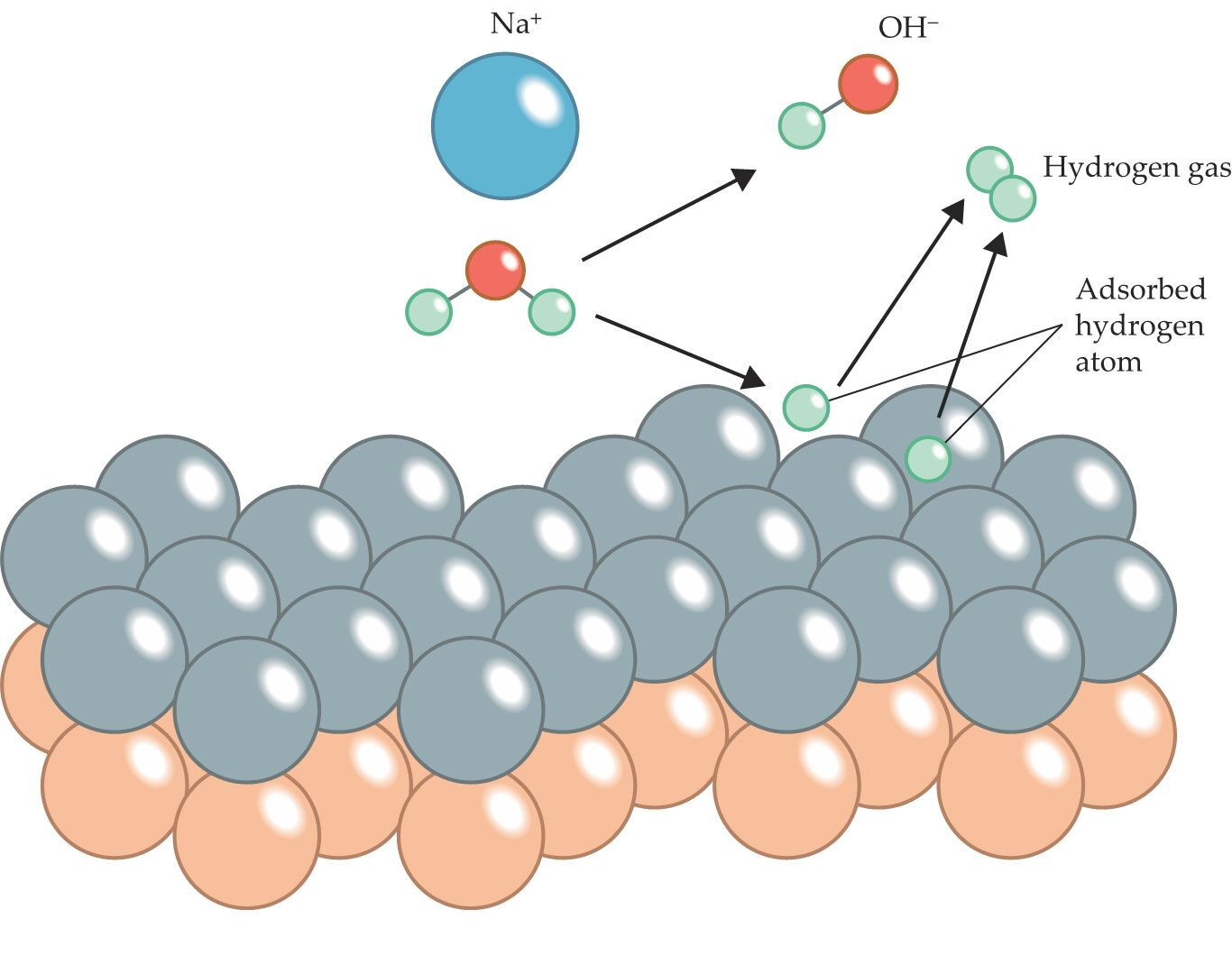
At the water–electrode interface, the researchers applied an electric potential ranging from 0.29 V to −1.11 V. Figure
Figure 2.

Raman shifts help reveal the underlying structure of water in an aqueous electrocatalytic system. The spectral peak centered at approximately 550 cm−1 grows increasingly prominent at progressively more negative electric potentials. The peak corresponds to a vibrational motion of water whose molecules have formed a series of densely packed layers near an electrode surface. The Raman shift at approximately 3400 cm−1 indicates three modes of water’s oxygen–hydrogen bond (red, orange, and blue peaks). At highly negative electric potentials, the O–H bonds with weakened hydrogen bonding and strongly hydrated sodium ions (red peaks) prevail over the O–H bonds with fully filled hydrogen bonds (blue peaks). (Adapted from ref.
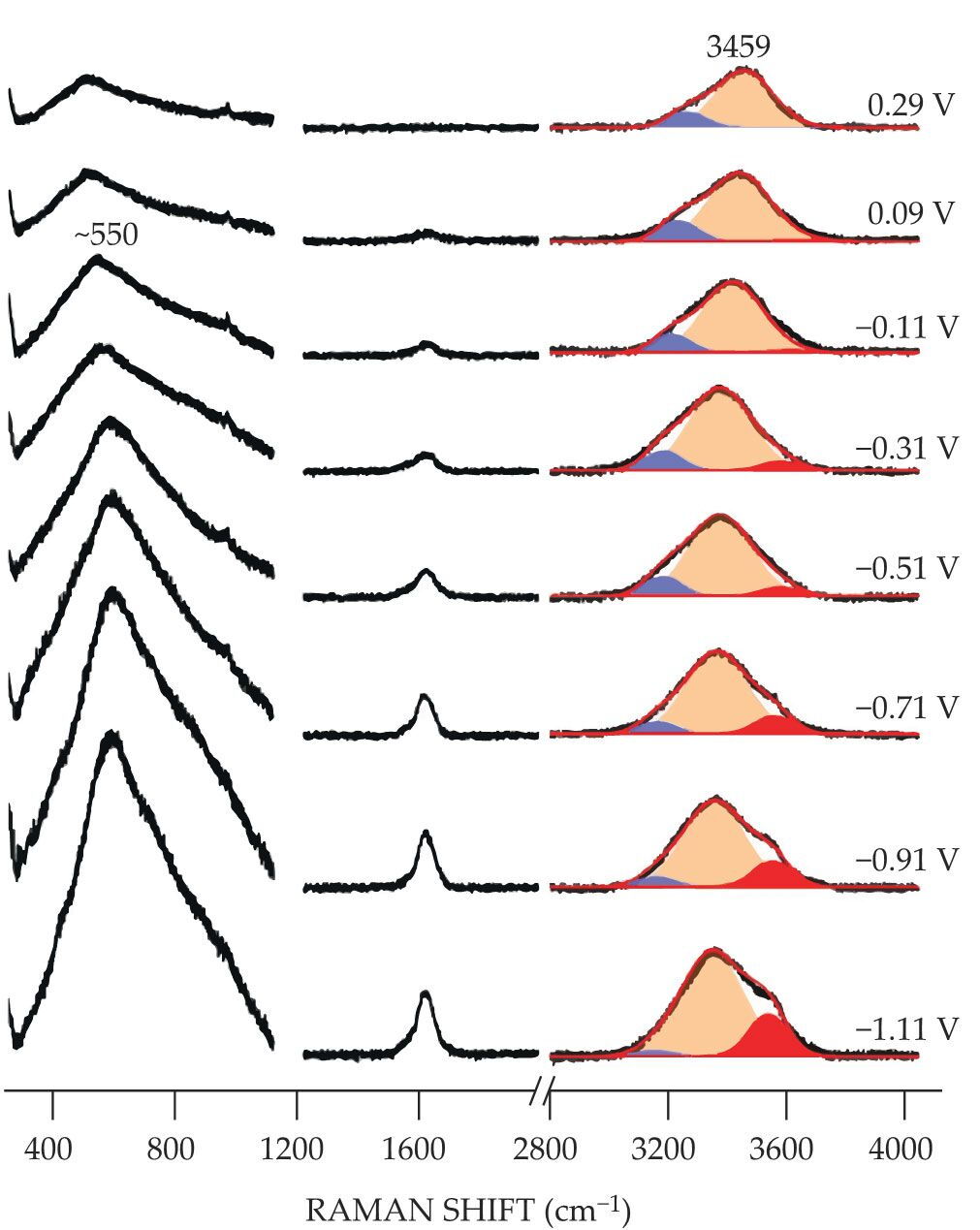
The intensity of the 550 cm−1 band steadily increased as the electric potential became more negative. The new results agree with previous work that found the 550 cm−1 band is related to an ordered water structure. 3 At a distance of up to three molecular diameters from the electrode, water forms layers of molecules far denser, and consequently with different properties, than bulk water.
A structure revealed
To learn more about how sodium contributes to the ordered structure of interfacial water, Pan and some of his colleagues modeled the experimental system using density functional theory. Their ab initio molecular dynamics simulations found that sodium weakens the network of hydrogen bonds that help hold water molecules together.
The enfeebled hydrogen bonding is also evident in the spectral observations of the oxygen–hydrogen bond, shown in figure
The liberated individual water molecules gather in a ring around a sodium ion to form a hydration shell. The positively charged hydrated sodium is naturally attracted to the negatively charged surface of the palladium–gold electrode. In fact, the simulations show that hydrated sodium brings water with weakened hydrogen bonds nearer to the electrode surface than it would otherwise reach without the sodium.
Once sufficiently close to the electrode, some hydrogen preferentially adsorbs to the surface, and Pd–H bonds form. After a series of additional chemical steps, H2 gas is produced.
According to the simulations, Pd–H bonds are shorter at progressively more negative potentials and at higher concentrations of hydrated sodium. A shorter bond is a stronger one and, therefore, promotes more water dissociation. The proximity of water molecules to the metal surface, the researchers say, also improves the electron-transfer efficiency between the electrode and the interfacial water. Under those conditions, the hydrogen-evolution reaction can proceed more easily and efficiently.
The researchers note that the relevance of sodium cations in improving the production rate of H2 may be applicable for cations in other aqueous electrocatalytic reactions. Converting carbon dioxide to hydrocarbons or nitrogen to ammonia, for example, requires water dissociation. Tuning cations to more carefully control the structure of interfacial water may help improve the rate and efficiency of those reactions too.
References
1. Y.-H. Wang et al., Nature 600, 81 (2021). https://doi.org/10.1038/s41586-021-04068-z
2. J.-F. Li et al., Nature 464, 392 (2010). https://doi.org/10.1038/nature08907
3. M. F. Toney et al., Nature 368, 444 (1994). https://doi.org/10.1038/368444a0
More about the authors
Alex Lopatka, alopatka@aip.org





{kind=link}
{kind=link}