Supercooled and Glassy Water
DOI: 10.1063/1.1595053
One of the four Aristotelian elements, water has played a central role in scientific thought for millennia. 1 To the physical scientist it is a continuing source of fascination because of its many unusual and counterintuitive properties. For example, liquid water, if sufficiently cold, expands and becomes more compressible when cooled, and less viscous when compressed. Water can also exist in at least two distinct glass forms–a phenomenon known as polyamorphism.
Water is not only fascinating, but it is also one of the most important and ubiquitous substances on Earth. There are 1.3 × 109 km3 of water in the oceans, 3.3 × 107 km3 in the polar ice caps, 2 × 105 km3 in glaciers, 105 km3 in lakes, and 1.2 × 103 km3 in rivers. In addition, 2.2 × 105 km3 of water fall annually as precipitation. 1 Nearly every aspect of our daily lives is influenced or controlled by water. From agriculture to travel, and from public health to commerce, the properties of water shape human activity and define the geography, topography, and environment in which we live. Indeed, life itself cannot exist without water.
Water can exist in many different crystalline forms, 13 of which have been identified to date. Of those, nine are stable over some range of temperature and pressure–for example, at atmospheric pressure, ordinary hexagonal ice is stable between 72 and 273 K–and the other forms are metastable. 1 Although the stable form of water at sufficiently low temperature is invariably crystalline, liquid water can also exist inside the crystalline domain of stability. When that occurs, water is said to be supercooled.
Supercooled water exists in a state of precarious equilibrium. Minor perturbations such as dissolved or suspended impurities can trigger the sudden appearance of the stable crystalline phase. The largest natural inventory of supercooled water occurs in the form of small droplets in clouds and plays a key role in the processing of solar and terrestrial radiative energy fluxes. 1 Supercooled water is also important for life at subfreezing conditions, for the commercial preservation of proteins and cells, and for the prevention of hydrate formation in natural gas pipelines.
If liquid water is cooled fast enough (at rates on the order of 106 K/s), freezing can be avoided altogether, and water then becomes a noncrystalline solid–that is, a glass. Glassy water may be the most common form of water in the universe. It is observed as a frost on interstellar dust, constitutes the bulk of matter in comets, and is thought to play an important role in the phenomena associated with planetary activity. 2 Its formation in the laboratory, however, requires elaborate procedures.
The study of supercooled and glassy water is motivated not just by the ubiquity and technical relevance of H2O. Liquid water’s anomalies, though qualitatively significant, are quantitatively small at ambient temperature. But water’s odd behavior becomes much more pronounced at low temperatures. In fact, from a scientific perspective, water’s supercooled and glassy states pose some of the most interesting questions in contemporary condensed matter physics, such as whether a pure substance may have two critical points. This article discusses recent advances in our understanding of cold, noncrystalline water. A coherent interpretation of water’s properties is beginning to emerge as a result of experimental and theoretical investigations by chemists, physicists, and engineers. Nevertheless, key questions remain unanswered, and our goal is as much to summarize what is known as to underscore what remains to be understood.
Water’s anomalies
A salient characteristic of liquid water at atmospheric pressure is that its response functions (response of density ρ or entropy S to changes in temperature T or pressure P) increase sharply in magnitude upon cooling. As shown in figure 1, the increase begins at 319 K (46°C) for the isothermal compressibility KT ≡ (∂lnρ/∂lnP) T , 308 K (35°C) for the isobaric heat capacity CP ≡ T(∂S/∂T) P , and 277 K (4°C) for the magnitude of the coefficient of thermal expansion α P ≡ −(∂lnρ/∂T) P . Furthermore, the increases become much more pronounced the lower the temperature. Water’s behavior was carefully investigated in the 1970s by Austen Angell, now at Arizona State University, and his collaborators. 3 They measured KT down to 247 K. Fitting a number of thermophysical properties and their own KT measurements to power laws, they noted that thermodynamic response functions and characteristic relaxation times appear to diverge at a singular temperature T S = 228 K.

Figure 1. Water’s anomalies. Schematic dependence on temperature of (a) the isothermal compressibility KT , (b) the constant-pressure specific heat CP , and (c) the coefficient of thermal expansion α P . The behavior of water is indicated by the solid line; that of a typical liquid, by the dashed lines. Each of these three thermodynamic response functions is proportional to corresponding fluctuations in parameters such as the entropy or volume. The anomalous thermodynamics and fluctuations of liquid water are apparent above the melting temperature T m, and they become much more striking as one supercools below T m.

In 1982, Robin Speedy proposed that T S is associated with the spinodal curve–the line where superheated water becomes intrinsically unstable with respect to vapor formation. 4 In a normal liquid at sufficiently low temperatures, the limit of stability with respect to boiling occurs at negative pressures (that is, the liquid is under tension–see the article by Humphrey Maris and Sebastien Balibar in Physics Today, February 2000, page 29 ). According to Speedy’s interpretation, in water at low enough temperatures, the limit of stability instead reverts to positive pressure. It is now understood that thermodynamic consistency arguments combined with simulation results do not support the existence of a spinodal singularity at 228 K: A retracing spinodal would require that the metastable continuation of water’s boiling curve terminate at a critical point below the triple point temperature. Furthermore, because of homogeneous nucleation of ice–formation without assistance from impurities or surfaces–measurements cannot be made close enough to the presumed singularity to warrant reliable power-law fits such as those used by Angell and his collaborators. Indeed, the smallest reduced temperature T/T S − 1 at which measurements have been made is about 0.03, compared to 10−3 or better in state-of-the-art critical phenomena experiments. Angell’s work nevertheless sparked enormous interest in understanding the behavior of cold water, an interest that continues undiminished.
Each response function is associated with a microscopic fluctuation. For instance, the isothermal compressibility is proportional to volume fluctuations: kTVKT = 〈(δV) 2 〉, where V is the mean value of the fluctuating volume for a fixed number of molecules, k is Boltzmann’s constant, and δ denotes a fluctuation in a quantity about its mean value. Similarly, the isobaric heat capacity is proportional to the entropy fluctuations at fixed pressure: kCP = 〈(δS)2〉. The thermal expansion coefficient reflects the cross-correlations between entropy and volume fluctuations, since kTVαP = 〈δ SδV〉.
Figure 1 illustrates the striking contrast between water and typical liquids, for which density and entropy fluctuations become smaller as the temperature decreases. In water, density and entropy fluctuations become more pronounced the lower the temperature. Volume and entropy fluctuations in most liquids are positively correlated: An increase in volume results in a corresponding increase in entropy. In water below 277 K, volume and entropy fluctuations are anticorrelated: An increase in volume brings about a decrease in entropy. Such anomalies are apparent in stable liquid water and become increasingly pronounced when one supercools water.
The microscopic cause of the observed anticorrelation between volume and entropy fluctuations is the tetrahedral symmetry of the local order around each water molecule. 5 As water is cooled, the closest neighbors begin to order, gradually taking on the local four-coordinated geometry appropriate for the structure of the water molecule, with its two positively charged lobes containing the protons and with its two lone pairs of electrons. A noncovalent interaction between an electropositive hydrogen atom on one molecule and an electronegative oxygen atom on another molecule is called a hydrogen bond. In water, hydrogen bonds favor local tetrahedral symmetry. Thus, in ordinary ice, each water molecule has four nearest neighbors and acts as a hydrogen donor to two of them and as a hydrogen acceptor from the other two. These nearest neighbors are located near the vertices of a regular tetrahedron surrounding the central oxygen atom. The H-O-H bond angle of an isolated water molecule, 104.5°, is in fact very close to the tetrahedral angle, 109.5°.
Whereas ice is a permanent tetrahedral network held together by hydrogen bonds, liquid water’s tetrahedrality is local and transient. Regions of local tetrahedral order possess a larger specific volume than the average–unlike regions of, say, local close-packed order. The entropy, on the other hand, always decreases upon cooling, because the specific heat is, of necessity, positive. As temperature decreases, the local specific volume increases due to the progressive increase in tetrahedral order. Thus the entropy and volume can become anticorrelated, and αP can become negative. Other liquids, such as silica, that possess local tetrahedral symmetry but do not have hydrogen bonds display the same property.
Figure 2a shows a picture of a transient cluster taken from a simulation of liquid water. Two molecules belong to the same cluster if they possess a high enough degree of local tetrahedral symmetry (as measured by the relative positions of a molecule’s oxygen atom and those of its four nearest neighbors) and if their oxygen atoms are sufficiently close. Although all molecules possess high local tetrahedral symmetry with respect to their four nearest neighbors, not every such neighbor belongs to the cluster. The connectivity in the cluster is imposed by hydrogen bonds, which can be seen in a hydrogen atom of one molecule pointing toward the oxygen atom of a neighboring molecule. The mean volume per molecule in this type of cluster is larger than that of the bulk. 6

Figure 2. Static and dynamic heterogeneities play important roles in the properties of supercooled water. (a) Instantaneous configuration of a 15-molecule cluster taken from a Monte Carlo simulation of supercooled water at 240 K. Oxygen atoms are represented by red spheres, and hydrogen atoms by white spheres. Tetrahedral symmetry can be seen in the fourth molecule starting from the cluster’s left end and in the sixth molecule starting from the right end. The cluster, which has a lower density than the mean value for the entire system, is stabilized by highly directional and noncovalent hydrogen bonds between hydrogen and oxygen atoms on different molecules. (b) Dynamic cluster composed of 16 “mobile molecules” (defined as the 7% of the molecules that move most during a 2-picosecond time interval) found in molecular dynamics simulations of supercooled water at 260 K. Rather than being uniformly distributed, mobile molecules tend to be found in clusters, as indicated by the yellow lines. As the temperature decreases, both static and dynamic clusters increase in size; the precise characterization of these static and dynamic heterogeneities (such as their shape, size, and lifetime) is an active area of current research.
(Adapted from ref. 6.)

Dynamical effects
If liquid water is sufficiently cold, its diffusivity increases and its viscosity decreases upon compression. Pressure disrupts the tetrahedral hydrogen-bond network, and the molecular mobility consequently increases. In contrast, compression of most other liquids leads to a progressive loss of fluidity as molecules are squeezed closer together. The anomalous pressure dependence of water’s transport coefficients 1 occurs below about 283 K for the diffusivity and below about 306 K for the viscosity, and persists up to pressures of around 2 kbar. One qualitative physical explanation for this anomalous pressure dependence is Le Chatelier’s principle: When a thermodynamic system is at equilibrium and external conditions are altered, the equilibrium will adjust so as to oppose the imposed change. In water, the large-volume clusters (such as the one in figure 2a) that become significant at low temperature will reduce in size and number under pressure, so water will become more like a normal liquid.
Recent molecular dynamics simulations of diffusion show that as the temperature is lowered into the supercooled region, motion becomes increasingly complex. During a randomly selected picosecond time interval in low-temperature simulations, most of the water molecules are not translating; instead, they are confined or “caged” by the hydrogen-bonded network. A small fraction of the molecules, however, are breaking out of their cages. Rather than being isolated, these newly freed molecules appear to form clusters (figure 2b) not altogether unlike the dynamic heterogeneities that are believed to be distinguishing features of supercooled liquids in general. 6 Thus supercooled water is both spatially (figure 2a) and dynamically (figure 2b) heterogeneous.
Ranges of stability
Important temperature ranges for water at atmospheric pressure are shown in figure 3. Water, like any liquid, can be heated above its boiling point without undergoing a phase transition. The attainable degree of such superheating is controlled by the rate of nucleation, and is about 553 K at atmospheric pressure, 180 K above the boiling point. Kinetics also controls the attainable extent of supercooling. At atmospheric pressure, it is possible to supercool water to its homogeneous nucleation temperature T H ≈ 231 K, at which the nucleation rate suddenly becomes very large–for example, 1015 nuclei/(cm3·s). Thus, the temperature range over which water can exist as a liquid is more than three times larger than the normal stability range (273–373 K). 1

Figure 3. Temperature ranges for liquid water at atmospheric pressure. Equilibrium (thermodynamic) transitions are indicated by solid lines, and kinetic ones by dotted lines. The limits of superheating and supercooling correspond to homogeneous nucleation of the vapor and crystal phases, respectively, in small but macroscopic liquid droplets (about 3 µm in diameter) devoid of impurities. The temperature range between the superheating and supercooling limits is more than three times wider than the stable liquid range. What is presumably highly viscous water above the glass transition T g can be studied between T g and approximately 150 K, where crystallization to cubic ice (ice Ic, one of ice’s 13 crystal structures) occurs. It is also possible to observe liquid water below the supercooling “limit” by evaporative cooling of small clusters. Experiments between 136 and 231 K are frontier areas of current research. So too is the precise location of the glass transition: Recent research suggests that water’s true glass transition would occur at 165 K if crystallization to cubic ice did not intervene.
(Courtesy of O. Mishima.)
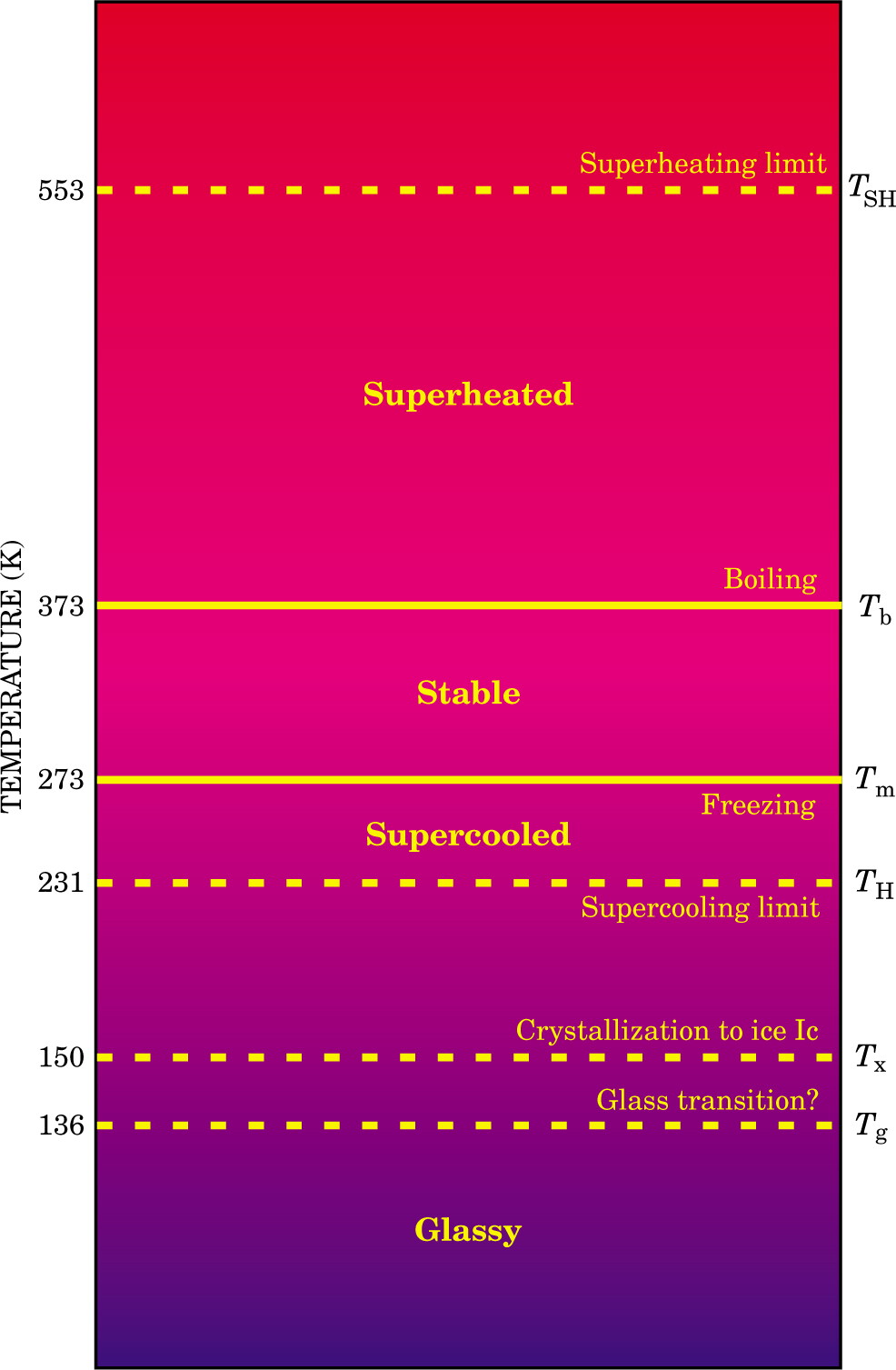
Limits of supercooling and superheating, being kinetic in nature, are not absolute. They can be bypassed provided that the observation time τobs is shorter than the nucleation time τnucl (about 10 µs for a 3-µm-diameter droplet close to T H). If, in addition, the thermal equilibration time τequil is shorter than the observation time (τequil ≪τobs ≪τnucl), then measurements are meaningful from a thermodynamic point of view, because the sample attains metastable equilibrium in times much shorter than those required to make measurements. 1
The interval between TH and the glass transition temperature Tg is a frontier domain whose experimental exploration is key to a full understanding of metastable water. The direct observation of supercooled water in this experimental range is challenging regardless of whether one attempts to enter this “no man’s land” by cooling liquid water or by heating glassy water. Supercooling is challenging because the nucleation time becomes extremely short below the homogeneous nucleation temperature T H. Hence, to bypass crystallization, water needs to be cooled extremely fast, and real-time observations are therefore challenging. In the 140–150 K range, water’s extremely large viscosity causes the nucleation rate to slow down, which in principle allows for much longer observation times despite the very large degree of supercooling. However, if one heats glassy water, it crystallizes to cubic ice at about 150 K. Promising forays into water’s no man’s land include temperature-programmed desorption measurements of isotope exchange and mixing rates on thin amorphous water films. 7
Glassy water
Glasses are nonequilibrium materials, so their physical properties depend on the process used to make them. Thus it is not surprising that different forms of glassy water can be obtained by following different experimental protocols. However, water is very unusual in that the transformation between different forms can be sharp and reversible and is accompanied by large changes in fundamental physical properties, such as the density. Such behavior is suggestive of a thermodynamic phase transition, and the quest for understanding the nature of such a transition, if one truly exists, underlies much of the current interest in glassy water.
Two forms of glassy water have been extensively studied: low-density and high-density amorphous ice (LDA and HDA, respectively). 8–11 Recently, very-high-density amorphous ice (VHDA) has been proposed as a new, distinct form of glassy water. 12 The glassy states differ in structure, as measured by neutron diffraction, x-ray diffraction, and Raman spectroscopy, and in bulk properties, such as density. The experimental paths to glassy water and the transitions between its different forms are illustrated in figure 4.

Figure 4. Polyamorphism is the term for having multiple distinct glassy states. (a) Routes to the formation of low-density (LDA), high-density (HDA), and very-high-density (VHDA) amorphous ice. LDA is formed by rapid cooling of water vapor or liquid water, after annealing. It is also formed by heating decompressed HDA or VHDA. HDA is formed by pressure-induced amorphization of ordinary ice (ice Ih), compression of LDA, rapid cooling of emulsified liquid water at high pressure, or constant-volume (isochoric) heating of VHDA. VHDA is formed by annealing HDA at high pressure. All of these processes are irreversible. (b) Reversible transformation between LDA and HDA by pressure cycling at about 135 K and 2 kbar. (c) The glass transition of LDA is conventionally assigned a temperature Tg of 136 K. Increasing the temperature leads to the formation of very viscous liquid water and crystallization to cubic ice at 150 K. An alternative suggestion is that Tg should be 165 K.

Glassy water was first produced in the laboratory in 1935 by Eli Burton and W. F. Oliver at the University of Toronto, who deposited water vapor onto a cold metal plate. 8 Nowadays, thin films of vapor-deposited glassy water are typically grown, using molecular beams, on single-crystal substrates at deposition rates between 0.1 and 7 µm/h and temperatures between 10 and 120 K. The direct vitrification of liquid water was first achieved in 1980 by Peter Brüggeller and Erwin Mayer at the University of Innsbruck; 9 they directed a pressurized jet of micron-sized water droplets in n-heptane onto liquid ethane at 90 K. Using rapid cooling of small aerosolized water droplets on a metal cryoplate cooled below 77 K, Mayer and his collaborators subsequently vitrified pure liquid water without resorting to a liquid cryomedium. Vitrification of water by rapid cooling of the liquid is referred to as hyperquenching, because it requires cooling rates of about 106 K/s. Both vapor-deposited and hyperquenched samples, after annealing, relax to LDA. In 1984, Osamu Mishima (now at the National Institute for Materials Science in Tsukuba, Japan) and his coworkers compressed ordinary ice at 77 K to about 11 kbar and formed HDA by pressure-induced amorphization. 10 At 77 K, decompressed HDA does not relax to its expanded state, but when heated to around 120 K, it forms LDA. The transition from one form of amorphous water to the other is abrupt, with about a 25% change in density, and can be reversed. 11
The commonly accepted value for water’s glass transition temperature at ambient pressure is T g = 136 K. Studying the thermal behavior of hyperquenched glassy water when it is heated toward its glass transition, Angell and coworkers recently noted that the release of heat (enthalpy relaxation) does not occur until the sample is anomalously close to its glass transition temperature. 13 Such behavior is contrary to what is observed in many other glass-forming liquids, in which thermal relaxation begins at lower values of T/T. Normal behavior is restored if water’s Tg is reassigned a value of 165 K, which Angell and his collaborators argue should be water’s true glass transition temperature. This reassignment is controversial because spontaneous crystallization to cubic ice at around 150 K precludes direct observation of this higher Tg. The debate is forcing a useful reexamination of important experiments, including the reversible cycling between LDA and HDA.
Developing a coherent picture
A coherent picture of the thermodynamics of metastable water should explain the following:
-
▸ the sharp increase in isothermal compressibility, the isobaric heat capacity, and the magnitude of the thermal expansion coefficient upon supercooling;
-
▸ the nature of the transition between LDA and HDA; and
-
▸ the relationship between supercooled and glassy water. The known experimental observations can be rationalized in two ways, known as the liquid–liquid phase transition 14 and singularity-free 15 hypotheses (see figure 5).

Figure 5. Two thermodynamic scenarios have been proposed for the phase behavior of metastable water. In the liquid–liquid phase transition hypothesis, a first-order phase transition (red line) occurs between two distinct forms of supercooled liquid water: low-density liquid (LDL) and high-density liquid (HDL). The transition between high-density amorphous ice (HDA) and low-density amorphous ice (LDA) is the low-temperature manifestation of that phase change; physical properties change smoothly in going from LDL to LDA and from HDL to HDA. The first-order transition terminates at a critical point, C’. (C is the liquid–gas critical point; the liquid–gas coexistence curve is shown in green.) Because C’ falls between the homogeneous nucleation curve (T H) and the crystallization curve to cubic ice (T X), it is extremely challenging to observe experimentally. In the singularity-free scenario, there is no coexistence of different phases at equilibrium. Instead, the transformation from LDL to HDL is continuous and occurs across a narrow but finite region (blue) of temperatures and pressures. In both scenarios, the character of the liquid (or amorphous) phase changes upon cooling at sufficiently high pressure, as the dense, high-entropy phase transforms to the less dense and more ordered phase.
View larger ![]()


According to the liquid–liquid phase transition hypothesis, the transition between LDA and HDA is a low-temperature manifestation of a first-order transition between two phases of liquid water: low-density liquid (LDL) and high-density liquid (HDL); LDA and HDA are simply the corresponding vitreous forms. The transition terminates at a liquid–liquid critical point; at higher temperatures, the HDL and LDL phases are indistinguishable, just as the gas and liquid phases are indistinguishable above a liquid–gas critical point. Associated with any critical point are “critical fluctuations” that are pronounced well above the critical temperature, so the hypothesized liquid–liquid critical point explains the dramatic increase in quantities such as the compressibility, specific heat, and thermal expansion coefficient. Theoretical and computational estimates locate the liquid–liquid critical point below the homogeneous nucleation temperature; the critical point is thus difficult to probe experimentally because of rapid crystallization. The exothermic nature of the changes involved in going from HDA to LDA implies that HDA has a greater entropy. According to the Clausius–Clapeyron relation, which connects the slope of the coexistence curve to the entropy and volume change of the phase transition, a transition in which the denser phase is more disordered has a coexistence line with a negative slope in the P–T plane. The second, liquid–liquid critical point thus would occur at the low-pressure, high-temperature end of the LDA–HDA equilibrium locus.
Experimental evidence in support of the existence of a liquid–liquid phase transition in water has recently been found. 16 If the melting line of ice IV, a metastable phase of crystalline water, intersects the hypothesized LDL–HDL transition line, the liquid in equilibrium with ice IV will change its entropy and specific volume abruptly. Consequently, by the Clausius–Clapeyron relation, the slope of the melting curve will also change abruptly. In 1998, Mishima and one of us (Stanley) measured the melting line of ice IV and observed a sharp change in its slope; that finding has since been confirmed in heavy water and repeated for the melting line of ice V. Water’s homogeneous nucleation locus lies very close to the hypothesized LDL–HDL transition line, and it is difficult to distinguish discontinuous changes in the slope of a melting line (as required by the liquid–liquid phase transition hypothesis) from sharp but continuous ones, so the experiments are less conclusive than desired. Hence, Mishima and Stanley used experimental data to calculate the Gibbs potential. Along the melting line of each ice polymorph, the Gibbs potential of the liquid must equal the Gibbs potential of the solid. Because there are many ice polymorphs, and thus many melting lines, one can measure the Gibbs potential of liquid water at a sufficient number of state points to permit estimation, by interpolation, of the entire function. The estimated function displays a liquid–liquid critical point at a pressure of about 1 kbar and a temperature of 220 K.
In the singularity-free scenario, 15 the amorphous states are again the vitreous forms of LDL and HDL. Upon supercooling, the response functions increase sharply but remain finite: They display pronounced maxima with respect to temperature but they do not diverge. Accordingly, the transition between LDA and HDA is continuous. Because, by definition, sharp maxima in the response functions imply large changes in entropy and volume, the transition between LDA and HDA is predicted to occur in a narrow interval of temperature and pressure that is difficult to distinguish experimentally from a true line when glassy phases are involved (see figure 5). In this viewpoint, the increase in response functions upon supercooling is not a reflection of an underlying singularity but the inevitable consequence of the existence of a line along which water’s thermal expansion coefficient vanishes. In the singularity-free scenario, the fluctuations between LDL and HDL remain finite, and the predicted density and enthalpy changes along any thermodynamic path remain continuous. Recent neutron and x-ray diffraction measurements of structural changes during the LDA–HDA transition have been interpreted as suggesting the existence of multiple distinct amorphous forms, 17 consistent with the possibility of a continuous transition.
In both scenarios, the amorphous states are smoothly connected to the liquid state. In the liquid–liquid phase transition picture, LDL is smoothly connected to LDA, HDL to HDA, and at sufficiently low temperatures and high pressures, discontinuous LDA–HDA transitions occur. In the singularity-free picture, the LDL–LDA and HDL–HDA connections are also smooth, but no discontinuity exists between LDA and HDA. The continuity of states between supercooled and glassy water has been verified by calorimetry, neutron diffraction, and computer simulation.
The structure of VHDA has been determined recently by isotope substitution neutron diffraction. 12 VHDA’s overall significance with respect to metastable water’s phase behavior is not yet understood. In particular, it is not known with certainty whether VHDA is a distinct phase, separated from HDA by a first-order transition, or whether it is simply very dense HDA.
Modeling water
Underlying cold water’s oddities are microscopic fluctuations between dense, disordered, high-energy local configurations and comparatively more ordered, low-energy open ones. The liquid–liquid phase transition and singularity-free scenarios differ only in their predictions regarding the magnitude of these fluctuations. Nevertheless, the shape and temporal stability of the “ordered’” (LDA-like) and “disordered” (HDA-like) domains in liquid water is unknown at present. This question is ideally suited to computational scrutiny, and a number of simulations are under way.
By revealing how the time-averaged local structure around a water molecule changes with pressure, recent neutron diffraction experiments at 268 K support the notion of fluctuations between low- and high-density local environments. 18 The experimentally measured structure of water can be expressed as a linear combination of low- and high-density forms with a gradual transformation from low density to high density as the pressure increases. Figure 6 illustrates these structures, obtained by computer simulation, in the form of three-dimensional contours of constant density around a central water molecule. The inner shell (labeled I in the figure) shows the spatial distribution of tetrahedrally bonded molecules. The second shell (II), in antiphase with the first shell, collapses at high pressure. This collapse is the primary signature of the structural transformation that occurs as density increases.

Figure 6. Contours of constant density around a central water molecule at 268 K for (a) low-density liquid water (LDL) and (b) high-density liquid water (HDL). The oxygen atom is shown in red; hydrogen atoms are partially visible as white lobes attached to the oxygen. The contours were calculated by a computer simulation that is constrained to reproduce the experimentally determined parameters (namely, the OO, OH, and HH structure factors measured by neutron diffraction
(Courtesy of A. K. Soper and M. A. Ricci.)

The behavior of liquid water has intrigued physical scientists for centuries. The quest to unravel water’s many mysteries impacts chemistry, biology, geology, materials science, engineering, and astronomy, as well as physics. This cross-fertilization is exemplified by the experiments on the liquid–liquid phase transition in phosphorus by Yoshinori Katayama and coworkers at the Japan Atomic Energy Research Institute. A vastly broadened perspective on the liquid state has resulted from research initially aimed primarily at understanding polyamorphism in water. Although a definitive picture of water’s low-temperature thermodynamics does not yet exist, the goal appears much closer now than it was a quarter century ago, when Angell called the attention of researchers to this fascinating problem in physics.
We thank Osamu Mishima and Francis Starr for helpful comments, and the Department of Energy and the National Science Foundation for support.
References
1. F. Franks, Water: A Matrix of Life, 2nd ed., Royal Society of Chemistry, Cambridge, UK (2000)
P. Ball, Life’s Matrix: A Biography of Water, Farrar, Straus, and Giroux, New York (2000)
O. Mishima, H. E. Stanley, Nature 396, 329 (1998) https://doi.org/10.1038/24540
P. G. Debenedetti, Metastable Liquids: Concepts and Principles, Princeton U. Press, Princeton, N.J. (1996).2. P. Jenniskens, D. F. Blake, Science 265, 753 (1994) https://doi.org/10.1126/science.11539186 .
3. R. J. Speedy, C. A. Angell, J. Chem. Phys. 65, 851 (1976) https://doi.org/10.1063/1.433153 .
4. R. J. Speedy, J. Phys. Chem. 86, 982 (1982) https://doi.org/10.1021/j100395a030 .
5. H. E. Stanley, J. Teixeira, J. Chem. Phys. 73, 3404 (1980) https://doi.org/10.1063/1.440538 .
6. J. R. Errington, P. G. Debenedetti, S. Torquato, Phys. Rev. Lett. 89, 215503 (2002) https://doi.org/10.1103/PhysRevLett.89.215503
N. Giovambattista, F. W. Starr, S. V. Buldyrev, H. E. Stanley, Phys. Rev. Lett. 90, 085506 (2003) https://doi.org/10.1103/PhysRevLett.90.085506 .7. R. S. Smith, B. D. Kay, Nature 398, 788 (1999) https://doi.org/10.1038/18601 .
8. E. F. Burton, W. F. Oliver, Proc. R. Soc. London, Ser. A 153, 166 (1935) https://doi.org/10.1098/rspa.1935.0229 .
9. P. Brüggeller, E. Mayer, Nature 288, 569 (1980) https://doi.org/10.1038/288569a0 .
10. O. Mishima, L. D. Calvert, E. Whalley, Nature 310, 393 (1984) https://doi.org/10.1038/310393a0 .
11. O. Mishima, J. Chem. Phys. 100, 5910 (1994) https://doi.org/10.1063/1.467103 .
12. T. Loerting, C. Salzmann, I. Kohl, E. Mayer, A. Hallbrucker, Phys. Chem. Chem. Phys. 3, 5355 (2001) https://doi.org/10.1039/b108676f
J. L. Finney, D. T. Bowron, A. K. Soper, T. Loerting, E. Mayer, A. Hallbrucker, Phys. Rev. Lett. 89, 205503 (2002) https://doi.org/10.1103/PhysRevLett.89.205503 .13. V. Velikov, S. Borick, C. A. Angell, Science 294, 2335 (2001) https://doi.org/10.1126/science.1061757 .
14. P. H. Poole, F. Sciortino, U. Essmann, H. E. Stanley, Nature 360, 324 (1992) https://doi.org/10.1038/360324a0 .
See also M.-C. Bellissent-Funel, Europhys. Lett. 42, 161 (1998) https://doi.org/10.1209/epl/i1998-00223-5 .15. S. Sastry, P. G. Debenedetti, F. Sciortino, H. E. Stanley, Phys. Rev. E 53, 6144 (1996) https://doi.org/10.1103/PhysRevE.53.6144 .
16. O. Mishima, H. E. Stanley, Nature 392, 164 (1998) https://doi.org/10.1038/32386
O. Mishima, Phys. Rev. Lett. 85, 334 (2000) https://doi.org/10.1103/PhysRevLett.85.334
O. Mishima, Y. Suzuki, Nature 419, 599 (2002) https://doi.org/10.1038/nature01106 .17. C. A. Tulk, C. J. Benmore, J. Urquidi, D. D. Klug, J. Neuefeind, B. Tomberli, P. A. Egelstaff, Science 297, 1320 (2002) https://doi.org/10.1126/science.1074178 .
See also I. Brovchenko, A. Geiger, A. Oleinikova, J. Chem. Phys. 118, 9473 (2003) https://doi.org/10.1063/1.1576372 .18. A. K. Soper, M. A. Ricci, Phys. Rev. Lett. 84, 2881 (2000) https://doi.org/10.1103/PhysRevLett.84.2881 .
More about the authors
Pablo Debenedetti (pdebene@princeton.edu ) is the Class of 1950 Professor of Engineering and Applied Science and chair of the department of chemical engineering at Princeton University. Gene Stanley (hes@bu.edu ) is University Professor, professor of physics and physiology, and director of the Center for Polymer Studies at Boston University.
Pablo G. Debenedetti, 1(pdebene@princeton.edu) Princeton University .
H. Eugene Stanley, 2(hes@bu.edu) Center for Polymer Studies, Boston University .





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}