Strongly Correlated Materials: Insights From Dynamical Mean-Field Theory
DOI: 10.1063/1.1712502
Modern solid-state physics explains the physical properties of numerous materials, such as simple metals and some semiconductors and insulators. But materials with open d and f electron shells, where electrons occupy narrow orbitals, have properties that are harder to explain. In transition metals, such as vanadium, iron, and their oxides, for example, electrons experience strong Coulombic repulsion because of their spatial confinement in those orbitals. Such strongly interacting or “correlated” electrons cannot be described as embedded in a static mean field generated by the other electrons. 1 The influence of an electron on the others is simply too pronounced for each to be treated independently.
The effect of correlations on materials properties is often profound. The interplay of the d and f electrons’ internal degrees of freedom—spin, charge, and orbital moment—can exhibit a whole zoo of exotic ordering phenomena at low temperatures. That interplay makes strongly correlated electron systems extremely sensitive to small changes in external parameters, such as temperature, pressure, or doping.
The dramatic effects can range from huge changes in the resistivity across the metal–insulator transition in vanadium oxide and considerable volume changes across phase transitions in actinides and lanthanides, to exceptionally high transition temperatures (above liquid-nitrogen temperatures) in superconductors with copper–oxygen planes. In materials called heavy fermion systems, mobile electrons at low temperature behave as if their masses were a thousand times the mass of a free electron in a simple metal. Some strongly correlated materials display a very large thermoelectric response; others, a great sensitivity to changes in an applied magnetic field—an effect dubbed colossal magnetoresistance. Such properties make the prospects for developing applications from correlated-electron materials exciting. But the richness of the phenomena, and the marked sensitivity to microscopic details, makes their experimental and analytical study all the more difficult.
The theoretical challenge
To understand materials made up of weakly correlated electrons—silicon or aluminum, for example—band theory, which imagines electrons behaving like extended plane waves, is a good starting point. That theory helps capture the delocalized nature of electrons in metals. Fermi liquid theory describes the transport of conduction electrons in momentum space and provides a simple but rigorous conceptual picture of the spectrum of excitations in a solid. In that description, excited states consist of independent quasiparticles that exist in a one-to-one correspondence to states in a reference system of noninteracting Fermi particles plus some additional collective modes.
To calculate the various microscopic properties of such solids, we have accurate quantitative techniques at our disposal. Density functional theory (DFT),
2
for example, allows us to compute the total energy of some materials with remarkable accuracy, starting merely from the atomic positions and charges of the atoms. See
However, the independent-electron model and the DFT method are not accurate enough when applied to strongly correlated materials. The failure of band theory was first noticed in insulators such as nickel oxide and manganese oxide, which have relatively low magnetic-ordering temperatures but large insulating gaps. Band theory incorrectly predicts them to be metallic when magnetic long-range order is absent.
Neville Mott showed that those insulators are better understood from a simple, real-space picture of the solid as a collection of localized electrons bound to atoms with open shells. Adding and removing electrons from an atom leaves it in an excited configuration. Because the internal degrees of freedom (like orbital angular momentum and spin) in the remaining atoms scatter the excited configurations, these states propagate through a crystal incoherently and broaden to form bands, called the lower and the upper Hubbard bands.
The modeling problem becomes more complicated as one moves away from these two well-understood, extreme limits and works with materials made up of electrons that are neither fully itinerant (propagating as Bloch waves in the crystal) nor fully localized on their atomic sites. The dual particle–wave character of the electron forces the adoption of components of the real-space and momentum-space pictures.
Systems with strongly correlated electrons fall within that middle ground. Traditionally, such materials have been described using the model Hamiltonian approach. That is, the Hamiltonian is simplified to take into account only a few relevant degrees of freedom—typically, the valence electron orbitals near the Fermi level. Reducing the full many-body Hamiltonian to a simpler, effective model retains the essence of the physical phenomena one wants to understand, but is itself a complicated problem.
One of the simplest models of correlated electrons is the Hubbard Hamiltonian, defined in equation 2(b) of

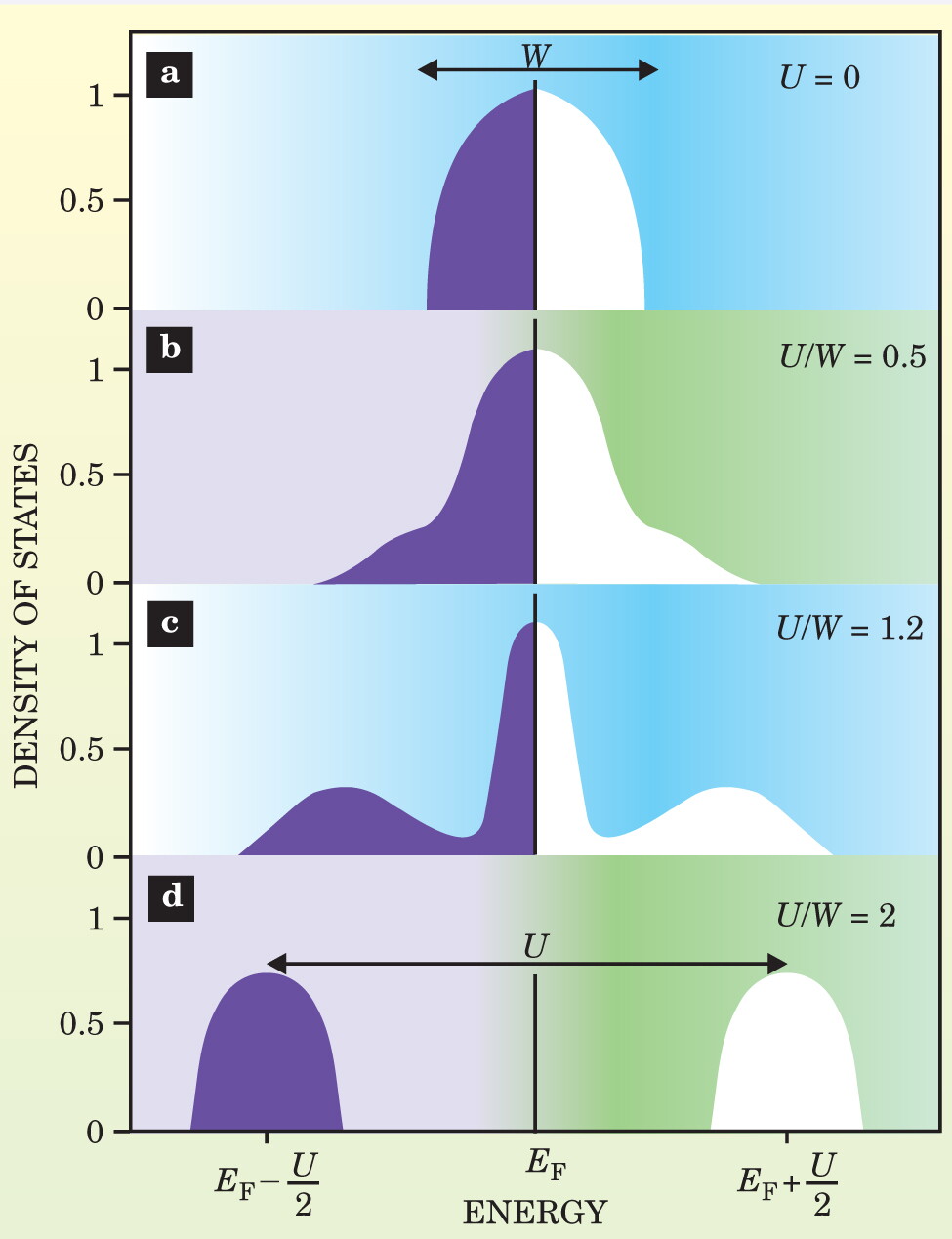
Figure 2. The density of states (DOS) of electrons in a material varies as a function of the local Coulomb interactions between them. This series illustrates how the spectral features—as measured by photoemission or tunneling experiments, say—evolve in the dynamical mean-field solution of the Hubbard model at zero temperature and half filling (the same number of electrons as lattice sites). U is the interaction energy and W is the bandwidth of noninteracting electrons. (a) For the case in which electrons are entirely independent, the DOS is assumed to have the form of a half ellipse with the Fermi level E F, located in the middle of the band, characteristic of a metal. (b) In the weakly correlated regime (small U), electrons can be described as quasiparticles whose DOS still resembles free electrons. The Fermi liquid model accounts for the narrowing of the peak. (c) In strongly correlated metals, the spectrum exhibits a characteristic three-peak structure: the Hubbard bands, which originate from local “atomic” excitations and are broadened by the hopping of electrons away from the atom, and the quasiparticle peak near the Fermi level. (d) The Mott metal–insulator transition occurs when the electron interactions are sufficiently strong to cause the quasiparticle peak to vanish as the spectral weight of that low-frequency peak is transferred to the high-frequency Hubbard bands.
The lattice structure and hopping terms influence the ability of the electrons to order magnetically, especially in the insulating state. If the magnetic ordering is impeded—crucial for observing subtle transitions between nonmagnetic phases—the system is said to be “frustrated.”
Despite using model Hamiltonians to simplify the problem, even simple properties such as the phase diagram of a strongly correlated electron system are difficult to calculate exactly. Given the rich variety of physical properties in such systems, though, it is important to develop techniques that are tractable and yet remain flexible enough to allow theorists to incorporate material-specific details into the calculations. Dynamical mean-field theory (DMFT) has been allowing researchers to make strides in that direction.
Dynamical mean-field theory
A wide variety of numerical techniques and analytical methods have been used to treat strongly correlated electron systems. In 1989, Walter Metzner and one of us (Vollhardt) introduced a new limit to the correlated electron problem, that of infinite lattice coordination: Each lattice site is imagined to have infinitely many neighbors. 3 That approach retained the competition between kinetic energy and Coulomb interaction of electrons while simplifying the computation. And it triggered a multitude of investigations of the Hubbard and related models taken in this limit. Consequently, a better understanding of various approximation schemes emerged along with an exact solution of some simpler models. 4
A second advance came when Antoine Georges and one of us (Kotliar) mapped the Hubbard model (a lattice model) onto a self-consistent quantum impurity model—a set of local quantum mechanical degrees of freedom that interacts with a bath or continuum of noninteracting excitations. 5 (For a subsequent significant derivation, see Mark Jarrell’s article in ref. .) That construction provides the basis of the dynamical mean-field theory of correlated electrons. 7 It allowed many-body theorists to formulate and solve a variety of model Hamiltonians on the lattice using analytic 5 and numerical techniques such as quantum Monte Carlo, 6 previously developed to study impurity models. 7 The DMFT solutions become exact as the number of neighbors increases.
In essence, a mean-field theory reduces (or maps) a many-body lattice problem to a single-site problem with effective parameters. Consider the classical theory of magnetism as an analogy: Spin is the relevant degree of freedom at a single site and the medium is represented by an effective magnetic field (the classical mean field). In the fermionic case, the degrees of freedom at a single site are the quantum states of the atom inside a selected central unit cell of the crystal; the rest of the crystal is described as a reservoir of noninteracting electrons that can be emitted or absorbed in the atom. Figure 1 depicts that emission or absorption as mediated by a quantum mechanical amplitude V ν , and

Figure 1. Dynamical mean-field theory (DMFT) of correlated-electron solids replaces the full lattice of atoms and electrons with a single impurity atom imagined to exist in a bath of electrons. The approximation captures the dynamics of electrons on a central atom (in orange) as it fluctuates among different atomic configurations, shown here as snapshots in time. In the simplest case of an s orbital occupying an atom, fluctuations could vary among |0〉, |↑〉, |↓〉, or |↑ ↓〉, which refer to an unoccupied state, a state with a single electron of spin-up, one with spin-down, and a doubly occupied state with opposite spins. In this illustration of one possible sequence involving two transitions, an atom in an empty state absorbs an electron from the surrounding reservoir in each transition. The hybridization V ν is the quantum mechanical amplitude that specifies how likely a state flips between two different configurations.
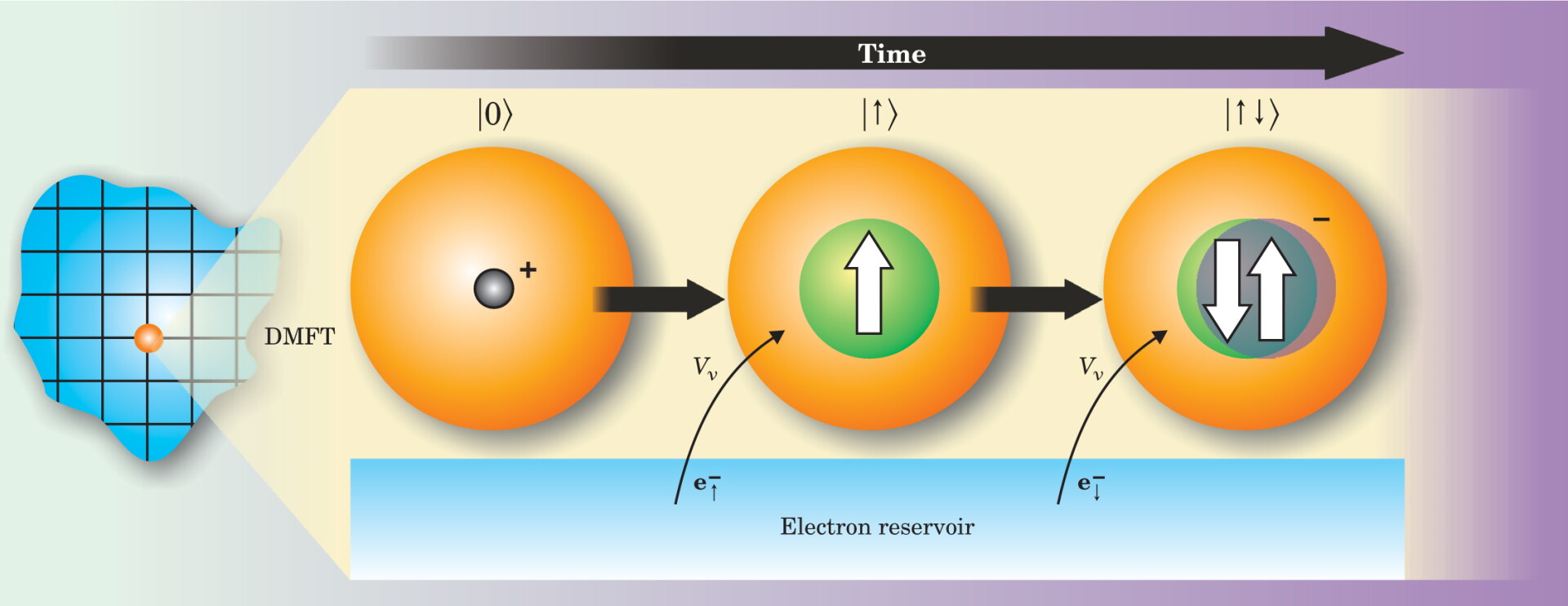
The local description of a correlated solid in terms of an atom embedded in a medium of noninteracting electrons corresponds to the celebrated Anderson impurity model, but now with an additional self-consistency condition. The hybridization function plays the role of a mean field and describes the ability of electrons to hop in and out of a given atomic site. When the hybridization is very small, the electron is almost entirely localized at a lattice site and moves only virtually, at short durations compatible with the Heisenberg uncertainty principle. On the other hand, when it is large, the electron can move throughout the crystal.
We thus obtain a simple local picture for the competition between itinerant and localized tendencies underlying the rich phenomena that correlated materials exhibit. The mapping of the lattice model onto an impurity model—the basis of the dynamical mean-field theory—simplifies the spatial dependence of the correlations among electrons and yet accounts fully for their dynamics—that is, the local quantum fluctuations missed in static mean-field treatments like the Hartree–Fock approximation.
Besides its conceptual value of providing a quantum analog of the classical mean field, the mapping of a lattice model onto the Anderson impurity model has had great practical impact. Applications of DMFT have led to a lot of progress in solving many of the problems inherent to strongly correlated electron systems, such as the Mott metal–insulator transition, doping of the Mott insulator, phase separation, and the competition of spin, charge, and orbital order.
The potential for system-specific modeling of materials using DMFT was recognized early on. 7 But actual implementation required combining ideas from band theory and many-body theory. 8 Vladimir Anisimov’s group in Ekaterinburg, Russia, and one of us (Kotliar) first demonstrated the feasibility of this approach using a simplified model of a doped three-dimensional Mott insulator, La1–x Sr x TiO3. The electrons in that material are divided into two sets: weakly correlated electrons, well described by a local-density approximation (LDA) that models the kinetic energy of electron hopping, and strongly correlated (or more localized) electrons—the titanium d orbitals—well described using DMFT. The one-body part of the Hamiltonian is derived from a so-called Kohn–Sham Hamiltonian. The on-site Coulomb interaction U is then added onto the heavy d and f orbitals to obtain a model Hamiltonian. DMFT (or more precisely, “LDA + DMFT”) is then used to solve that Hamiltonian.
Just as the Kohn–Sham equations serve as a reference system from which one can compute the exact density of a solid, the Anderson impurity model serves as a reference system from which one can extract the density of states of the strongly correlated electrons. The parallel between DFT and DMFT is best seen in the functional approach to DMFT,
9
outlined in
The metal–insulator transition
How does an electron change from itinerant to localized behavior within a solid when a control parameter such as pressure is varied? The question gets at the heart of the Mott transition problem, which occurs in materials as varied as vanadium oxide, nickel selenium sulfide, and layered organic compounds. 1,11 Those materials share similar high-temperature phase diagrams, despite having completely different crystal structures, orbital degeneracies, and band structures: In each, a first-order phase transition separates a high-conductivity phase from a high-resistivity phase. At low temperatures, in contrast, the materials exhibit very different ordered phases: Organics are superconducting, vanadium oxide forms a metallic and an antiferromagnetic insulating phase, and nickel selenium sulfide forms a broad region of itinerant antiferromagnetism.
The Mott transition is central to the problem of modeling strongly correlated electrons because it addresses directly the competition between kinetic energy and correlation energy—that is, the wavelike and particlelike character of electrons in the solid. Indeed, systems near the Mott transition display anomalous properties such as metallic conductivities smaller than the minimum predicted within a band picture, unconventional optical conductivities, and spectral functions outside of what band theory can describe.
1
A crucial question is how the local density of states—or more generally, the spectral function—varies as the ratio of the correlation strength U to bandwidth W increases (see figure
But how does the density of states evolve between these well-characterized limits? In DMFT, the spectral function of the electron contains both quasiparticle features and Hubbard bands in the intermediate correlation region. 5 A three-peak structure appears naturally within the Anderson impurity model, but was unexpected for a lattice model that describes the Mott transition. In fact, part of what makes DMFT an advance over previous techniques is that it allows researchers to obtain and equally treat the Hubbard bands and the quasiparticle peak within this three-peak structure. (DFT treats the quasiparticles alone, atomic theory treats the Hubbard band alone, but DMFT treats both.) Within DMFT, the Mott transition appears as the result of the transfer of spectral weight from the quasiparticle peak to the Hubbard bands of the correlated metallic state. 12 The transfer of spectral weight can be driven by pressure, doping, or temperature, and is responsible for the anomalous behavior near the Mott transition.
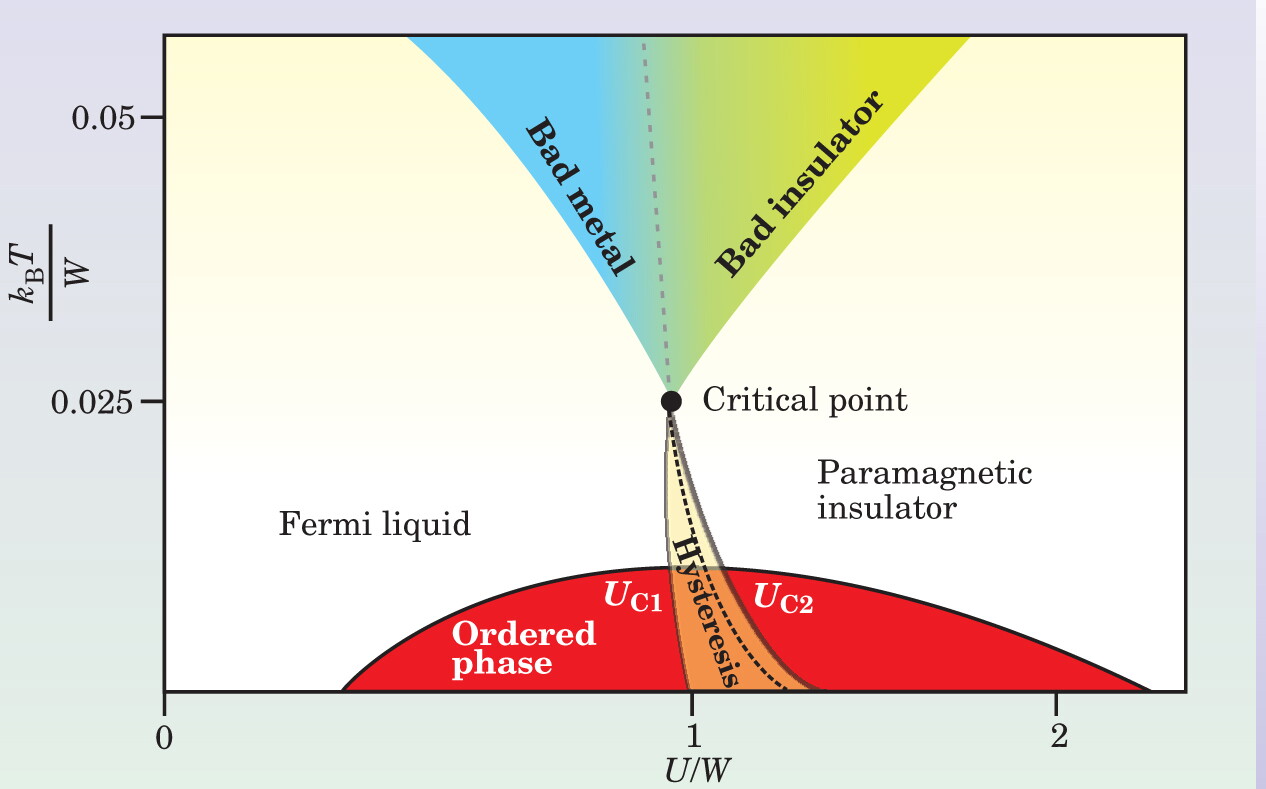
To appreciate the DMFT description of the Mott transition, consider the qualitative features of the phase diagram shown in figure 3. The figure essentially summarizes the features of a partially frustrated Hubbard model in different temperature regimes. The disappearance of metallic coherence and the closing of the high-energy Mott–Hubbard gap are distinct phenomena that occur in different regions of the phase diagram—at low temperatures in the form of lines at energies U C1 and U C2 that frame a hysteretic region, and at high temperature (above a second-order critical point) in the form of two distinct crossover lines that gradually separate metallic from insulating phases. 7

Figure 3. Schematic phase diagram of a material undergoing a Mott metal–insulator transition. Temperature and the strength of Coulombic repulsion U are plotted for a correlated electron material that is described using a Hubbard model. At low temperature, the system has long-range (for example, magnetic or orbital) order (red) according to solutions of the dynamical mean-field theory; the type of ordering is material- and model-dependent. In the orange region, the model has two distinct paramagnetic solutions bounded by the lines U C1 (where the insulator disappears) and U C2 (where the metal disappears). In equilibrium, a dotted first-order phase transition boundary indicates the position at which the free energy of these two solutions crosses. The first-order line terminates at a second-order critical point. At higher temperatures, the phase diagram is more universal. Systems as diverse as vanadium oxide alloys, nickel selenium sulfide, and organic materials exhibit the same qualitative behavior. Two crossover regimes indicate the change in materials properties as the electron interactions increase from left to right. The first illustrates the crossover from a Fermi liquid to a bad metal (blue), in which the resistivity is anomalously large. In the second crossover, materials take on the properties of a bad insulator (green), in which the resistivity decreases as temperature increases.
The Coulomb interactions and the matrix elements that describe electron hopping from site to site are the basic ingredients necessary to calculate the experimental phase diagram of materials near a Mott transition. The question is, Can the phase diagram be obtained from an electronic model alone, or is the coupling of electrons to the lattice required to obtain a first-order phase transition between a paramagnetic insulator and a paramagnetic metal? According to DMFT studies, an electronic model alone will do the job. Lattice changes across the phase transition are therefore the consequence, rather than the origin, of the discontinuous metal–insulator transition.
Now widely accepted, that conclusion was not reached without controversy. An analytic approach to the Mott transition was first required, 13 which deepened the analogy between the local spectral function and a statistical-mechanics order parameter. The electronic properties of the system—for example, the DC conductivity—behave like the order parameter of a liquid–gas transition or like the magnetization of the Ising spin model in statistical mechanics.
Experimental confirmation
Numerous efforts have been made to square the surprising aspects of the theory of the Mott transition with experimental observations. For instance, a collaboration of groups from Bell Labs and Rutgers University observed that pure vanadium oxide (V2O3) is located in the vicinity of the crossover line that continues the U C2 line (where the metallic phase disappears in figure 3) above the endpoint of the first-order transition line. 14 Using DMFT, this collaboration predicted that, with increasing temperature, resistivity would strongly increase and the weight of the quasiparticle peak would strongly decrease. And, indeed, optical conductivity measurements bore that prediction out. A group led by Gordon Thomas detected a transfer of optical intensity from low to high frequency with increasing temperature, a finding consistent with a temperature-dependent quasiparticle peak. 14
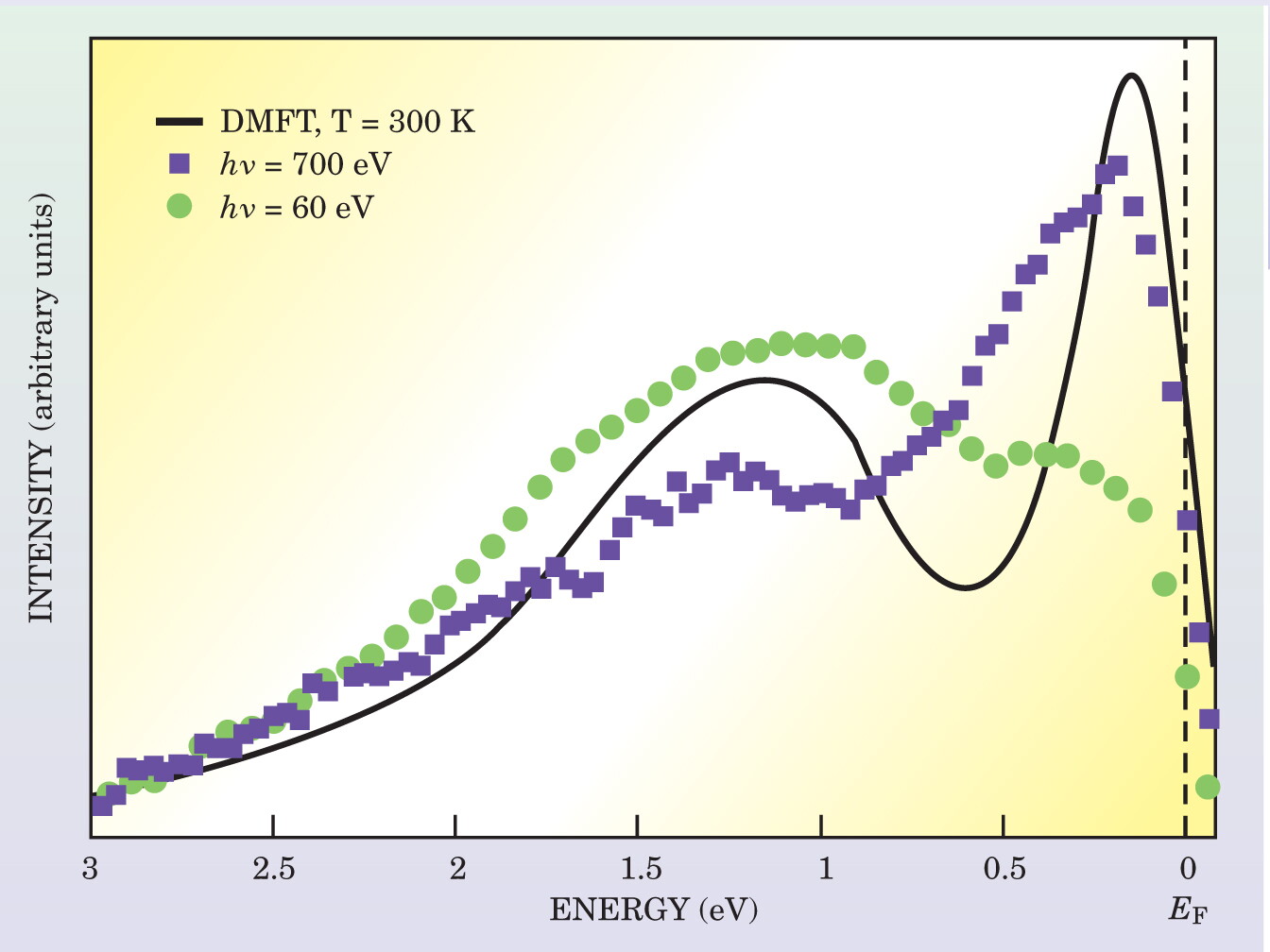
Only photoemission can measure the density of states of the correlated electrons directly. Z. X. Shen’s group at Stanford University observed a quasiparticle peak, well defined and separated from the Hubbard bands in nickel selenium sulfide and depending strongly on temperature and proximity to the Mott transition. More recently, an international collaboration of experimentalists and theorists observed well-defined and separated peaks in the metallic phase of vanadium oxide after first overcoming problems related to surface sensitivity that prevented clear observation in earlier studies (see figure 4). 15

Figure 4. Photoemission spectrum of metallic vanadium oxide (V2O3) near the metal–insulator transition. The dynamical mean-field theory calculation (solid curve) mimics the qualitative features of the experimental spectra. The theory resolves the sharp quasiparticle band adjacent to the Fermi level and the occupied Hubbard band, which accounts for the effect of localized d electrons in the lattice. Higher-energy photons (used to create the blue spectrum) are less surface sensitive and can better resolve the quasiparticle peak.
(Adapted from Mo et al., ref. 15.)
In the Sr1–x Ca x VO3 system, researchers observed a similar quasiparticle peak well separated from the Hubbard band. The quasiparticle signal decreased in intensity with increasing calcium doping, x. Attempts to interpret the spectra within DMFT as enhanced electron correlation with increasing x, however, did not square with other experimental results; the specific heat, for instance, was measured as essentially independent of x. A group led by D. D. Sarma and Isao Inoue and one by Shigemasa Suga helped resolve the puzzle by performing bulk-sensitive photoemission experiments. 16 Earlier photoemission experiments had given the false impression that the electronic structure was evolving toward a more correlated system as doping increased, when, in fact, only the surface states were doing so. On the theoretical side, first-principles calculations demonstrated that the strength of the correlations do not significantly depend on the concentration x.
This kind of successful blending of DMFT and experiment is also occurring in studies of electronic transport. In materials such as vanadium oxide, nickel selenium sulfide, and κ-organics, researchers have been able to map theoretically the various regimes of the phase diagrams in some detail. 11,1 After many unsuccessful attempts, even the critical behavior predicted by the theory has been observed in transport measurements around the critical endpoint of vanadium oxide and organic materials. 17
Other materials
The application of DMFT concepts and techniques to a broad range of materials and a wide variety of strong-correlation problems is now an active research frontier. The materials and topics under study include manganites, ruthenates, vanadates, actinides, lanthanides, fullerenes, quantum criticality in heavy fermion systems, magnetic semiconductors, and Bechgaard salts (quasi-1D organic materials). A deeper understanding of the metal–insulator transition is useful not just for predicting the properties of that list of materials, but provides insights into diverse electronic structure problems. Plutonium and cerium, for instance, exhibit itinerant magnetism and suffer a large change in the crystal volume at the Mott transition. The following brief case studies outline the relevance of DMFT to these elements.
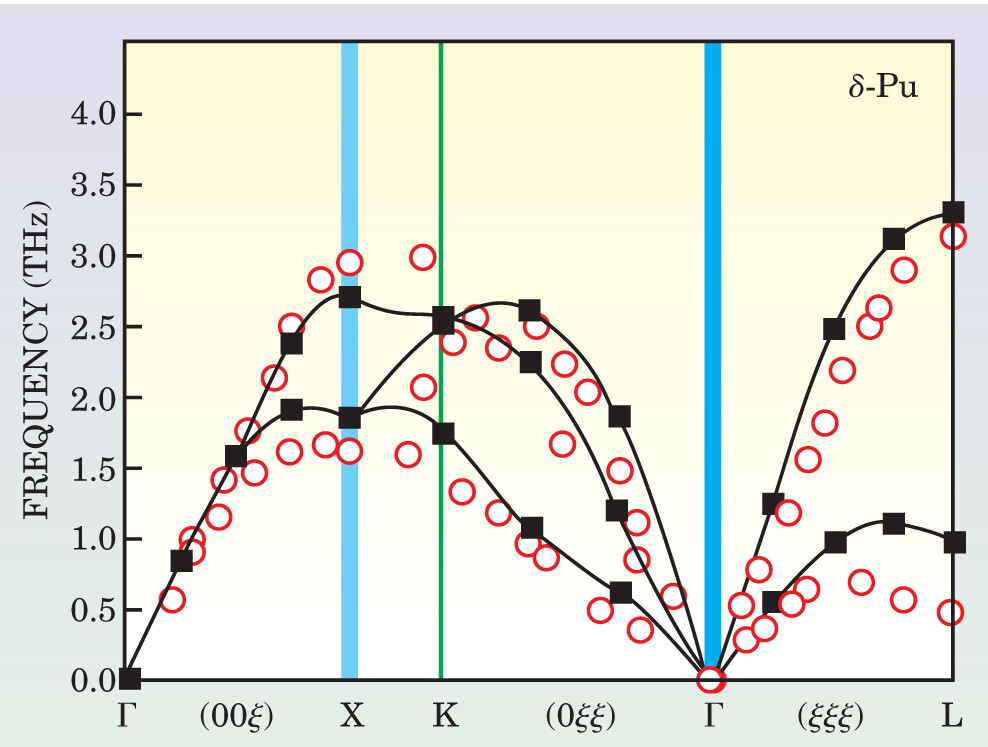
Plutonium , in its stable δ phase, is not well described within LDA. That approximation underestimates the phase volume by nearly 30% and predicts a nonexistent unstable phase. Using DMFT, researchers realized that the plutonium f electrons stabilize the material near a Mott transition, and they were able to interpret calculated photoemission spectra in terms of the quasiparticle peak and Hubbard bands described previously. More recently, a collaboration among Rutgers University, New Jersey Institute of Technology, and Los Alamos National Laboratory predicted the theoretical spectrum of vibrations of δ-Pu using DMFT. Figure 5 illustrates the remarkable agreement between the theoretical vibrational spectrum and recent experimental measurements. 18

Figure 5. Predicted theoretical phonon spectrum of plutonium (red), calculated using dynamical mean-field theory, compares well with the experimental values (solid squares), measured by inelastic x-ray scattering. The dispersion curves plotted here map the vibrational frequencies of the atoms in momentum space. The frequencies vary with the wave vectors and are evident in the branches as one moves in a particular Brillouin zone direction.
(Adapted from ref. 18.)
Cerium displays an isostructural transition 10 and also exhibits a large volume change (the so-called volume collapse) between two of its phases, α and γ. Jim Allen and Richard Martin, then at Xerox Corp, interpreted the thermodynamics and spectral changes across the α–γ transition in terms of the Anderson impurity model more than 20 years ago. Recently, Andy McMahan, Karsten Held, and Richard Scalettar calculated the spectra, total energies, and thermodynamics of cerium using DMFT 10 and found close accord with the experimental spectra. The calculation indicated a dramatic reduction of the 4f quasiparticle weight at the transition, responsible for the energy changes and the volume collapse at the transition.
Iron and nickel exhibit metallic ferromagnetism at low temperatures at which band theory applies. And a combination of DFT and LDA bears that out. However, at high temperatures, those elements behave more like a collection of atoms, described by a Curie susceptibility and a local magnetic moment reduced below the atomic value. DMFT provides a natural framework to describe both the high- and low-temperature regimes and the crossover between them. Indeed, in the case of nickel, Alexander Lichtenstein and collaborators found that the theoretical photoemission spectrum contains an experimentally well known satellite at energies well below the Fermi level. That feature, like the Hubbard band, is not reproduced in band theory. DMFT also predicts the correct magnetic properties above and below the Curie temperature.
Future directions
Judging by how reliably the theory reproduces complicated physical properties in a variety of materials, dynamical mean-field methods clearly represent a new advance in many-body physics. The strongly correlated electron regime of transition-metal oxides, for instance—in which electrons are neither fully itinerant nor localized—has simply not been accessible to other techniques.
Generalizations of the theory to an even wider variety of materials systems is an active area of research. By extending DMFT from single sites to clusters, for example, researchers can capture the effects of short-range correlations. And combining DMFT with advanced electronic-structure methods should make it possible to evaluate the Hamiltonian and the frequency-dependent screened Coulomb interaction from first principles without having to first construct a local density functional. 11 Similarly, combining realistic DMFT total energies (calculated as a function of atomic positions) with molecular dynamics to treat the motion of ions and electrons simultaneously is another great challenge.
As the number of DMFT implementations and studies of new materials increases, more detailed comparisons with experiments are becoming possible. Such comparisons should help separate the effects that can be understood simply from local physics (captured by DMFT) from the effects that require long-wavelength modes not captured by the DMFT approach.
The DMFT equations have also been recast in a form useful for modeling strongly inhomogeneous materials, such as disordered alloys or interfaces—two new areas in which to explore strongly correlated phenomena. They are also areas of great technological importance. As growing computer power, novel algorithms, and new concepts bolster the ability of theorists to better model complicated systems, one can foresee a time when investigating correlated electron phenomena at nanoscales and in biological molecules is possible.
Density Functional Theory
In DFT, the basic quantity is the local electronic charge density of the solid, ρ(r). The total energy of the full, many-body problem of interacting quantum mechanical particles is expressed as a functional of this density:
The equation contains three parts: the kinetic energy of a noninteracting system T[ρ]; the potential energy of the crystal, V ext(r), plus the Hartree contribution to the Coulomb interaction between the charges; and the rest, denoted as the exchange and correlation energy term E xc. Minimizing the functional results in the Kohn–Sham equations
The Kohn–Sham equations serve as a reference system for DFT because they yield the correct ground-state density via
Dynamical Mean-Field Theory
To treat strongly correlated electrons, one has to introduce a frequency resolution for the electron occupancy at a particular lattice site. A Green function that specifies the probability amplitude required to create an electron with spin σ (↑ or ↓) at a site i at time τ′ and destroy it at the same site at a later time τ will do the job:
The Green function contains information about the local one-electron photoemission spectrum. The dynamical meanfield theory (DMFT) can be used to investigate the full many-body problem of interacting quantum mechanical particles or effective treatments such as the Hubbard model
1,7
The Anderson impurity model
By analogy with density functional theory, an exact functional of both the charge density and the local Green function of the correlated orbital is introduced as
The functional 9 has a similar decomposition as in DFT. However, the kinetic energy is no longer that of a free electron system because T [ρ, G] is the kinetic energy of a system with given density ρ(r) and local Green function G. DMFT provides an explicit approximation for E xc[ρ(r), G].
We are grateful to our research groups, collaborators, and colleagues for fruitful interactions; to NSF’s division of materials theory, the US Department of Energy, the Office of Naval Research, and the German Research Foundation (DFG) for financial support; and to the Aspen Center for Physics for hospitality.
References
1. For a recent review of the electronic correlation problem, see M. Imada, A. Fujimori, Y. Tokura, Rev. Mod. Phys. 70, 1039 (1998);https://doi.org/10.1103/RevModPhys.70.1039
for an early review of fermionic correlations, see D. Vollhardt, Rev. Mod. Phys. 56, 99 (1984).https://doi.org/10.1103/RevModPhys.56.992. R. R. Jones, O. Gunnarsson, Rev. Mod. Phys. 61, 689 (1989).https://doi.org/10.1103/RevModPhys.61.689
3. W. Metzner, D. Vollhardt, Phys. Rev. Lett. 62, 324 (1989).https://doi.org/10.1103/PhysRevLett.62.324
4. E. Müller-Hartmann, Z. Phys. B: Condens. Matter 74, 507 (1989);https://doi.org/10.1007/BF01311397
U. Brandt, C. Mielsch, Z. Phys. B: Condens. Matter 75, 365 (1989);https://doi.org/10.1007/BF01321824
V. Janiš, Z. Phys. B: Condens. Matter 83, 227 (1991).https://doi.org/10.1007/BF013094235. A. Georges, G. Kotliar, Phys. Rev. B 45, 6479 (1992).https://doi.org/10.1103/PhysRevB.45.6479
6. M. Jarrell, Phys. Rev. Lett. 69, 168 (1992);https://doi.org/10.1103/PhysRevLett.69.168
M. Rozenberg, X. X. Zhang, G. Kotliar, Phys. Rev. Lett. 69, 1236 (1992);https://doi.org/10.1103/PhysRevLett.69.1236
A. Georges, W. Krauth, Phys. Rev. Lett. 69, 1240 (1992).https://doi.org/10.1103/PhysRevLett.69.12407. For a general review of dynamical mean-field theory, see A. Georges, G. Kotliar, W. Krauth, M. Rozenberg, Rev. Mod. Phys. 68, 13 (1996); for a review of applications to the Hubbard model away from half filling, seehttps://doi.org/10.1103/RevModPhys.68.13
Th. Pruschke, M. Jarrell, J. J. Freericks, Adv. Phys. 44, 187 (1995).https://doi.org/10.1080/000187395001015268. V. V. Anisimov et al., J. Phys.: Condens. Matter 9, 7359 (1997);https://doi.org/10.1088/0953-8984/9/35/010
A. A. Lichtenstein, M. M. Katsnelson, Phys. Rev. B 57, 6884 (1998).https://doi.org/10.1103/PhysRevB.57.68849. R. Chitra, G. Kotliar, Phys. Rev. B 62, 12715 (2000);https://doi.org/10.1103/PhysRevB.62.12715
G. Kotliar, S. Savrasov, in New Theoretical Approaches to Strongly Correlated Systems, A. A. Tsvelik, ed., Kluwer Academic, New York (2001), available at http://arXiv.org/abs/cond-mat/0208241 .10. K. Held et al.,Psi-k Newsletter, April 2003, p. 65, available at http://psi-k.dl.ac.uk/newsletters/News_56/Highlight_56.pdf ;
A. A. Lichtenstein, M. M. Katsnelson, G. Kotliar, Electron Correlations and Materials Properties, 2nd ed., A. Gonis, N. Kioussis, M. Ciftan, ed. Kluwer Academic/Plenum, New York (2003), available at http://arXiv.org/abs/cond-mat/0211076 .11. Recent developments in the field are documented on the homepage of the Program on Realistic Theories of Correlated Electron Materials (2002) at the Kavli Institute for Theoretical Physics, Santa Barbara, CA. See http://online.kitp.ucsb.edu/online/cem02 .
12. A. Fujimori et al., Phys. Rev. Lett. 69, 1796 (1992);https://doi.org/10.1103/PhysRevLett.69.1796
X. X. Zhang, M. Rozenberg, G. Kotliar, Phys. Rev. Lett. 70, 1666 (1993);https://doi.org/10.1103/PhysRevLett.70.1666
A. Georges, W. Krauth, Phys. Rev. B 48, 7167 (1993).https://doi.org/10.1103/PhysRevB.48.716713. G. Kotliar, Eur. J. Phys. B 11, 27 (1999);https://doi.org/10.1007/s100510050914
G. Kotliar, E. Lange, M. Rozenberg, Phys. Rev. Lett. 84, 5180 (2000);https://doi.org/10.1103/PhysRevLett.84.5180
C. Castellani et al., Phys. Rev. Lett. 43, 1957 (1979).https://doi.org/10.1103/PhysRevLett.43.195714. M. Rozenberg et al., Phys. Rev. Lett. 75, 105 (1995).https://doi.org/10.1103/PhysRevLett.75.105
15. A. Matsuura et al., Phys. Rev. B 58, 3690 (1998);https://doi.org/10.1103/PhysRevB.58.3690
S.-K. Mo et al., Phys. Rev. Lett. 90, 186403 (2003).https://doi.org/10.1103/PhysRevLett.90.18640316. K. Maiti et al., Europhys. Lett. 55, 246 (2001);https://doi.org/10.1209/epl/i2001-00406-6
A. Sekiyama et al., http://arXiv.org/abs/cond-mat/0312429 .17. P. Limelette et al., Science 302, 89 (2003);https://doi.org/10.1126/science.1088386
F. Kagawa et al., http://arXiv.org/abs/cond-mat/0307304 .18. X. Dai et al., Science 300, 953 (2003);.https://doi.org/10.1126/science.1083428
J. Wong et al., Science 301, 1078 (2003).https://doi.org/10.1126/science.1087179
More about the authors
Gabriel Kotliar is a professor of physics at the Center for Materials Theory at Rutgers University in Piscataway, New Jersey.
Dieter Vollhardt is a professor at the Center for Electronic Correlations and Magnetism at the Institute for Physics, University of Augsburg in Germany.
Gabriel Kotliar, 1 Center for Materials Theory at Rutgers University in Piscataway, New Jersey, US .
Dieter Vollhardt, 2 Center for Electronic Correlations and Magnetism at the Institute for Physics, University of Augsburg in Germany .





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}