Record-breaking protein images have applications for drug discovery
DOI: 10.1063/PT.3.3254
Cancer is hard to treat. How is it possible to kill all the tumor cells when they’re spread throughout the body or buried deep inside an essential organ such as the brain? One possible strategy is to target the physical aspects of cancer, such as its rapid cell division (see Physics Today, August 2007, page 19 ) or the hastily grown, hole-riddled vessels that supply solid tumors with blood (see the article by Jennifer Grossman and Scott McNeil, Physics Today, August 2012, page 38 ).
Another approach is to identify and disrupt biochemical processes that occur only in cancer cells. Researchers are on the hunt for small-molecule drugs that can bind to the proteins involved in those processes and render them nonfunctional.
The search for the most effective drugs would be aided immeasurably by three-dimensional atomic-scale structures of those proteins and drug–protein complexes. But x-ray crystallography, the standard tool for finding molecular structures, isn’t always up to the job. Not all proteins can be easily crystallized, and even for those that can, the tightly packed crystal environment risks distorting their shape or changing how they interact with other molecules.
Now Sriram Subramaniam, of the National Institutes of Health in Bethesda, Maryland, and his colleagues have shown that cryoelectron microscopy may do the trick. 1 Cryo-EM has been used for decades to image biological nanostructures embedded in thin films of vitreous (that is, amorphous) ice, which is structurally similar to liquid water. (See the article by Bob Glaeser, Physics Today, January 2008, page 48 .) For most of that time, typical structural resolutions have hovered around 10 Å, which is nowhere near good enough to discern atomic bonds 1–1.5 Å long. And it’s been mostly limited to structures larger than 1000 kilodaltons (1 Da equals one atomic mass unit), which excludes most of the proteins relevant to the biology of cancer. Spurred by the recent advent of faster electron detectors, the field has been making rapid progress on both fronts. In their study of three cancer proteins, Subramaniam and colleagues are the first to achieve a resolution of better than 2 Å and to image a structure smaller than 100 kDa.
Thin ice
Transmission electron microscopy can image inorganic materials at atomic resolution with relative ease. An accelerated electron beam has a wavelength far smaller than any interatomic distance. And modern aberration-correction techniques allow the overall resolution to reach 0.5 Å (see the article by Yimei Zhu and Hermann Dürr, Physics Today, April 2015, page 32 ).
Organic and biological molecules, however, are more delicate than inorganic materials, and they can survive only a brief exposure to an electron beam without being destroyed. That exposure yields only a low-resolution image. To get a crisper picture, it’s necessary to spread the electron dose over tens of thousands of identical copies of the structure of interest and somehow combine the information gleaned from all of them into a single 3D structure. In the early years, researchers tried making 2D crystals with all the particles perfectly ordered. But since the late 1980s, most cryo-EM studies have used randomly oriented structures whose low-resolution images are then aligned computationally. That alignment is a lot easier for structures that are large, highly symmetric, or both: viruses, ribosomes, and multiprotein complexes.
For years, cryo-EM researchers have been faced with a tradeoff between two electron-detection techniques. Photographic film was the most efficient detector, but developing and digitizing it was a long and tedious process. On the other hand, CCD cameras, which convert incoming electrons to light before converting the light back into photoelectrons, offered automated detection in exchange for lower efficiency: With up to 90% of electrons going undetected, the individual images were fuzzier and harder to align.
That all changed with the development of direct electron detectors. Adapted from particle physics and commercialized by three different groups around the same time, they came into widespread use in cryo-EM in 2013. 2 The new detectors were not only both efficient and automated, they were also fast. For the first time, it was possible to subdivide a raw image, the product of a seconds-long exposure to the electron beam, into the frames of a miniature movie. By studying the data frame by frame, Nikolaus Grigorieff and colleagues discovered that the beam not only damaged individual molecules but also deformed the whole ice film—and the effect of the deformation was predictable. 3 By computationally undoing the deformation to realign the movie frames, they obtained much sharper images.
Resolution revolution
The field took off. Instead of the low-resolution structures that had earned cryo-EM the nickname “blob-ology,” researchers started getting structures that were tantalizingly close to showing atomic detail. And they started looking at single proteins, albeit large ones.
How much better could cryo-EM become? In 1995 Richard Henderson, one of the field’s pioneers, estimated that with perfect detectors, it should be possible to image 38 kDa molecules at 3 Å resolution. 4 Although that mass limit is still a long way off, several groups broke the 3 Å barrier last year. And the improvements kept coming.
The steady march toward ever better resolution—Subramaniam and colleagues’ new work included—is a product not of any further revolutionary changes to the technique but of the meticulous refinement of every aspect of the process. Cryo-EM structures are slow and laborious to obtain—Subramaniam estimates that the method is currently orders of magnitude more time consuming than x-ray crystallography—and many steps have yet to be fully optimized.
For example, one factor that influences image quality is the thickness of the ice film: Thinner films yield better images because electrons are less likely to scatter more than once. Because they were studying small proteins, Subramaniam and colleagues had the luxury of using thinner ice. But there’s no established method for precisely controlling ice thickness; instead, the NIH researchers had to prepare many specimens and focus their analysis on the ones that produced the best images.
Cancer proteins
Subramaniam and his team recorded cryo-EM structures of three proteins, all of which are relevant to cancer biology. Glutamate dehydrogenase (GDH), the largest of the three at 334 kDa, featured the record-breaking resolution of 1.8 Å. The researchers achieved 2.8 Å resolution of the 145 kDa lactate dehydrogenase (LDH), the enzyme responsible for producing lactic acid in sore, oxygen-starved muscles; cancer cells rely on the same reaction even when plenty of oxygen is available. And they obtained a 3.8-Å-resolution structure of isocitrate dehydrogenase (IDH), which at 93 kDa was the smallest protein yet resolved with cryo-EM. In many cancers, IDH contains a mutation that causes it to fuel malignant cell growth.
Figure
Figure 1. High-resolution images obtained with cryoelectron microscopy reveal the atomic structures of two proteins, lactate dehydrogenase (a) and glutamate dehydrogenase (b), as exemplified by the small portions of each protein shown here. The 1.8-Å-resolution glutamate dehydrogenase image, in particular, shows a level of atomic detail unprecedented in cryo-EM, including the carbon rings of the amino acids tryptophan, tyrosine, and phenylalanine, and some of the water molecules, shaded in yellow, that surround the protein. (Adapted from ref. Figure 2. The smallest protein imaged with cryoelectron microscopy, isocitrate dehydrogenase, both on its own (a) and bound to a molecular inhibitor known as ML309 (b). Although the 3.8-Å-resolution images fall short of showing atomic detail, the images offer new clues about how the molecules fit together. (Adapted from ref. 
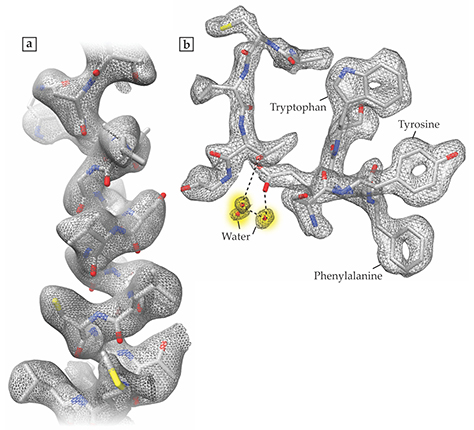

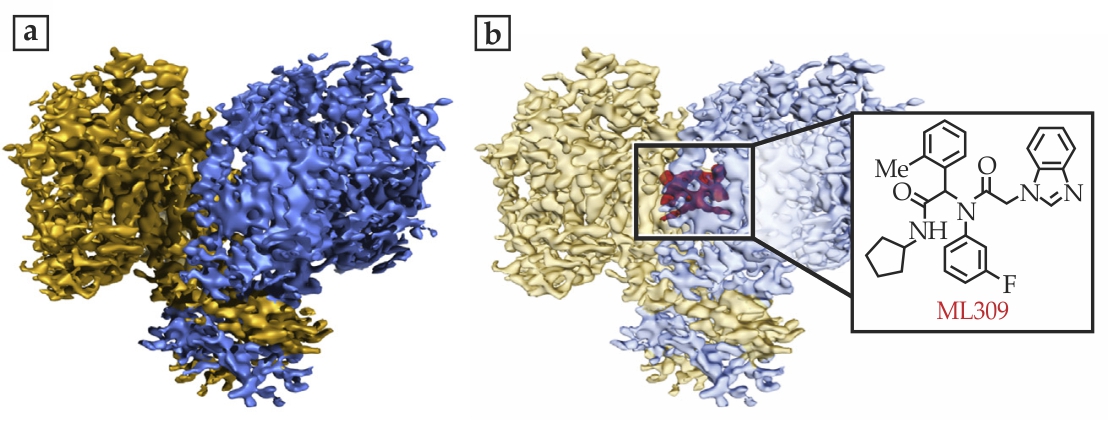
For the two smaller proteins, LDH and IDH, the researchers looked at the complexes formed between the biomolecules and small-molecule inhibitors that are currently under study as potential cancer-fighting drugs. Says Subramaniam, “We want to see how the molecules work, how they fit together.” Figure
A central remaining challenge for cryo-EM is that biomolecules with identical compositions don’t always have identical shapes: Even when folded into the correct structure, many proteins are floppy and can adopt multiple conformations in solution. The algorithms theorists have developed for image analysis 5 can deal with that structural diversity to a certain extent, but they still need to make assumptions in classifying and aligning the structural images. But cryo-EM will no doubt continue to improve as researchers test its ability to find structural information about the proteins involved in cancer, brain function, and other frontiers of biology.
References
1. A. Merk et al., Cell 165, 1698 (2016). https://doi.org/10.1016/j.cell.2016.05.040
2. X.-C. Bai et al., eLife 2, e00461 (2013);
X. Li et al., Nat. Methods 10, 584 (2013). https://doi.org/10.1038/nmeth.24723. A. F. Brilot et al., J. Struct. Biol. 177, 630 (2012). https://doi.org/10.1016/j.jsb.2012.02.003
4. R. Henderson, Q. Rev. Biophys. 28, 171 (1995). https://doi.org/10.1017/S003358350000305X
5. S. H. W. Scheres, J. Struct. Biol. 180, 519 (2012). https://doi.org/10.1016/j.jsb.2012.09.006
More about the authors
Johanna L. Miller, jmiller@aip.org





{kind=link}
{kind=link}