Chemistry Nobel honors mechanical bonds, molecular machines
DOI: 10.1063/PT.3.3382
Virtually all motion in biological systems, from the swimming of a bacterium to the beating of a blue whale’s heart, is initiated on a molecular level. Chemical fuel is consumed, and the energy that’s released, rather than being dispersed randomly into the environment, is channeled into specific modes of molecular movement. Flagella turn, molecules are shuttled across cell membranes, or motor proteins crawl along cytoskeletal filaments to initiate muscle contractions that perform macroscopic work.
Chemists are still a long way from creating synthetic molecules capable of mimicking those biological processes. But a series of breakthroughs by Jean-Pierre Sauvage, J. Fraser Stoddart, and Ben Feringa have provided researchers with components that may one day be assembled into larger molecular machines capable of performing complicated and useful tasks. Those breakthroughs have been honored by the 2016 Nobel Prize in Chemistry.
“There’s a tremendous potential for applications, yet unachieved, but mechanical chemistry has reached a certain maturity,” says R. Dean Astumian of the University of Maine. “We’ve got the simple machines—the wheels and the levers—and now it’s up to the creativity of the chemists to put them together.”
Molecules with moving parts are nothing new: Most chemical bonds can swivel, allowing part of the molecule to rotate with respect to the rest. Left to their own devices, though, most molecules either flop around at random or settle into a low-energy conformation if there is one. The challenge lies in controlling internal molecular motions to drive the system meaningfully away from equilibrium.
That challenge is compounded by the extremely low Reynolds number of the molecular environment. (See the article by Dean Astumian and Peter Hänggi, Physics Today, November 2002, page 33 .) To set a part of a macroscopic machine in motion, it’s often enough to give it a push and let Newtonian mechanics do the rest. But at the molecular scale, there is virtually no inertia, and machines are constantly batted around by the random thermal motion of the surrounding molecules—the so-called Brownian storm. Molecular machines are operated not by applying forces and torques but through engineering their energy landscape.
Chains and wheels
For the most part, organic molecules are held together by the attractive forces manifested by covalent chemical bonds. But many of today’s molecular machines are based on a fundamentally different kind of interaction, sometimes called a mechanical bond: the interlocking of molecular components that cannot separate because of the repulsive forces that prevent covalent bonds from passing through each other. Mechanically interlocked molecules include catenanes, or sets of linked rings, and rotaxanes, or rings threaded onto dumbbell-shaped rods.
Chemists’ curiosity about catenanes and rotaxanes dates back more than 60 years, piqued in part by the challenge of making the molecules. A blacksmith forging a macroscopic chain can make two open rings, thread one through the other, and close them up. But at the molecular scale, the process is not so straightforward. Although organic chemists had developed a rich toolbox of methods for making and breaking chemical bonds, there was no obvious way to control the positions of rings and axles that aren’t connected by any bonds.
Early synthesis methods fell into two classes, neither very effective. There were statistical methods, which in their simplest form involved making a large number of rings, then separating out the ones that happened to be in the right place at the right time to interlock. (Unsurprisingly, few were.) And there were scaffolded methods, which entailed holding the partial rings in place with a temporary network of organic bonds, completing the rings, and then breaking down the temporary bonds. That approach offered greater control than the statistical methods did, but it was tremendously complicated, often requiring 20 steps or more. Because each of those steps resulted in some loss of product, overall yields were too small for chemists to begin to explore any uses for the unusual molecules.
In 1983 Sauvage came up with a different approach: using a copper ion to temporarily hold the rings in place. 1 As chemists already knew, ions of copper and other metals can form diverse compounds by surrounding themselves with an array of molecules called ligands. Importantly, each metal–ligand bond is formed by a pair of electrons both donated by the ligand—as opposed to one electron from each bonded atom, as is typical for a covalent bond. As a result, it’s possible in many cases to cleave the bond without affecting the ligand’s stability as a molecule.
Sauvage used Cu+ ions, which tend to form tetrahedral complexes with four ligands. But the ligands don’t need to all be separate; it’s also possible for the ion to bond with multiple atoms of the same ligand. Sauvage realized that he could create a complex with two curved molecules, each with two bonds to the central Cu+ ion, as shown in figure 1a. The molecules would be held perpendicular to each other, in prime position to be built up into a set of interlocked rings. Removing the ion is somewhat more difficult than dissociating a normal metal-ion complex, because the rings block the ion’s escape. But it can be done, with a good yield, in a single step.

Figure 1. Mechanically interlocked molecules. (a) A set of two linked molecular rings called a catenane, from the Latin for “chain,” was first synthesized efficiently with the help of a copper ion (green) to temporarily hold the partially formed links together. (b) A rotaxane, from the Latin for “wheel” and “axle,” can be switched between two configurations by changing the chemical environment. (c) A daisy-chain molecule, consisting of two interconnected rotaxane switches, changes in length when the rotaxanes are rearranged. (Panels b and c courtesy of Edie Sevick.)
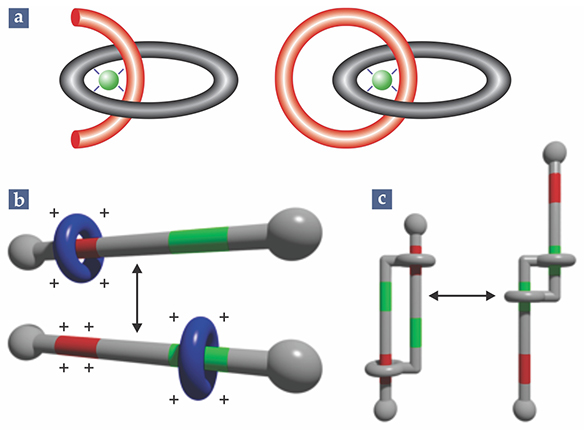
Because their components are free to move, mechanically interlocked molecules can rearrange, or isomerize, in ways that ordinary molecules cannot. In particular, the ring of a rotaxane can not only rotate but also translate. To make operable molecular machines, researchers would still need to find a way to control those new degrees of freedom. Stoddart achieved just that in 1994, when he developed the first rotaxane switch. 2
The key features of Stoddart’s switch are the two chemically distinct “docking sites” on the axle, shown in red and green in figure 1b. Normally, the ring has an affinity for the red site. But when the red site is endowed with a positive charge, either by adding reagents or passing a current through the solution, the ring, also positively charged, is shuttled to the green site. The process is fully reversible: Remove the charge, and the ring returns to the red site.
Rotaxane switches gave rise to other molecular configurations, such as the daisy-chain molecule in figure 1c, first synthesized in 2000 by Sauvage. 3 Because switching the interlinked rotaxanes actually changes the molecule’s length, it became easier to envision molecular machines performing work. Indeed, Sauvage’s paper was titled, “Towards synthetic molecular muscles.”
One-way rotors
Feringa expanded the range of controllable molecular movements by creating molecular motors that could spin in a desired direction, an essential property for a molecular engine. Rather than mechanically interlocked molecules, the motors are based on so-called overcrowded alkenes. 4 An alkene’s defining feature, the carbon–carbon double bond, is normally both rigid and planar. Making the alkenes “overcrowded” are the large lobe-like chemical groups attached to each of the two central carbons: They’re so big and bulky that they can’t all sit in a single plane simultaneously. The molecule must therefore twist slightly, which gives rise to the four distinct stable or metastable isomers shown schematically in figure 2.

Figure 2. A molecular motor based on a central carbon–carbon double bond. Applied heat and UV light cause the motor to cycle through its four distinct isomers shown here. Thanks to additional asymmetry (not represented in this schematic) of the outer lobelike chemical groups, the rotation proceeds in one direction only.
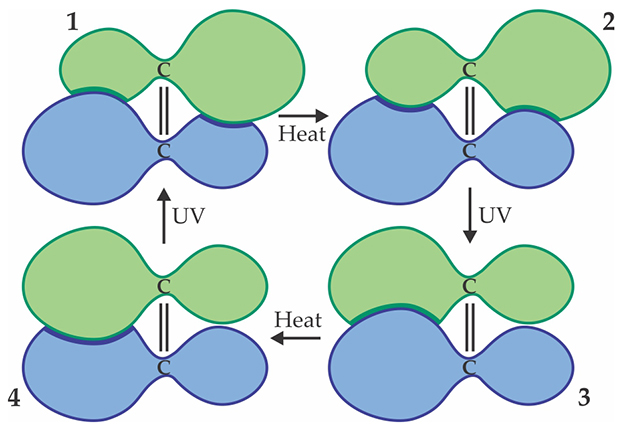
At first it might seem thermodynamically impossible to make such a molecule rotate only in one direction. One of the four isomers must be the lowest in energy, and any attempt to prod the molecule toward a higher-energy configuration would be equally likely to twist it in either direction. Feringa achieved one-way rotation by exploiting molecular asymmetries and two sources of energy.
The transitions from isomer 1 to 2 and from 3 to 4 each keep the double bond locked in place while the outer lobes slip past each other; their energy barriers are low enough that they can be surmounted thermally. On the other hand, the transitions from 2 to 3 and 4 to 1, which require the double bond to flip-flop, must be achieved photochemically: UV excitation temporarily displaces the electrons responsible for keeping the bond planar, so the molecule can swivel.
Feringa designed the lobes to be slightly asymmetric about the plane of the page, so that isomers 1 and 2 aren’t mirror images of each other (which would necessarily have the same energy) but different structures with distinct energies, with 2 lower in energy than 1. Likewise, isomer 4 is lower in energy than 3, so the molecule was thermodynamically guided in the right direction. For the photochemical steps, he took advantage of differences in the molecules’ UV absorption spectra by choosing a wavelength at which isomers 2 and 4 absorb strongly but 1 and 3 do not.
For the 1999 proof-of-principle experiment, Feringa applied the energy stimuli sequentially: cooling the molecules to −55 °C before irradiating them with UV, then warming the solution to room temperature or above while the UV light was turned off. That allowed him to examine the molecular structure after each step and make sure the motors were spinning the way they should. But the process works equally well when heat and light are applied simultaneously, which makes the motors rotate continuously.
Building blocks
Stoddart’s first rotaxane switch had to be chilled to −44 °C; at higher temperatures, thermal energy would rip the ring away from its preferred docking site, and distinct translational isomers could not be resolved. And Feringa’s first molecular motor took hours to complete a single rotation at room temperature. That sluggishness was due entirely to the time it took to surmount the substantial thermal barriers—the photochemical steps, in contrast, were accomplished in picoseconds.
Tinkering with the chemistry brought better performance. Rotaxane docking sites with different molecular designs interact more strongly with the threaded rings and offer good selectivity even at room temperature. And changing the size and shape of a rotor’s lobes adjusts its thermal barriers, which allows the rotation period to be tuned to any value between microseconds and years.
Researchers in the laureates’ groups and elsewhere are working on different ways to power the rudimentary molecular machines. There are now UV-triggered rotaxane switches, alkene rotors powered by visible light, rotors that get their energy from chemical reactions, and other variations. Chemically fueled machines are attractive because of their similarity to biomolecular machines. On the other hand, “Chemical fuels are often consumed inefficiently, and they produce a lot of waste products,” says Nathalie Katsonis of the University of Twente in the Netherlands. “With light, there is no waste, and that’s promising for a lot of applications. So it depends what you have in mind.”
As several groups have shown, it’s possible for molecular machinery to create motion on length scales much larger than the molecules themselves. A few examples are shown in figure 3. David Leigh (now at the University of Manchester in the UK) and colleagues used a surface covered with switchable rotaxanes to lift a macroscopic droplet of diiodomethane a short distance up a 12° incline: Changing the position of the rotaxane rings exposes and covers different chemical groups, which alters the wettability of the surface and causes the droplet to creep forward. 5 Feringa and colleagues used a liquid-crystal film doped with molecular rotors to spin a 28-µm-long glass rod. 6 And Katsonis and her colleagues created a polymer film with embedded photoswitchable molecules; when cut into ribbons, the film exhibits macroscopic motion in response to UV light. 7

Figure 3. Big movement from little molecules. (a) A droplet of diiodomethane is lifted a short distance up a 12° incline. The surface is lined with light-responsive rotaxanes that, when switched, increase the surface’s wettability. (Adapted from ref.
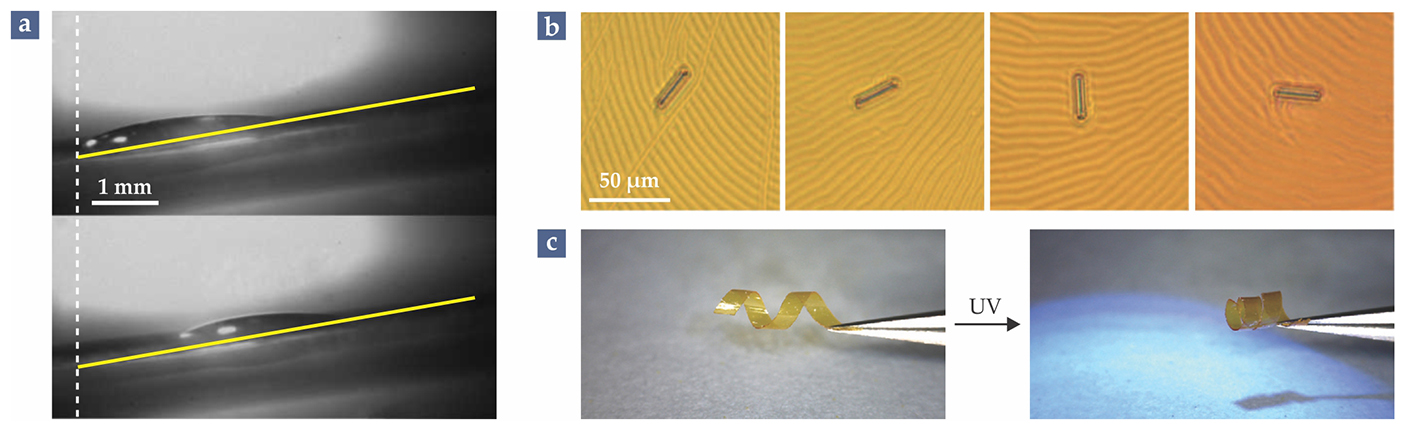
Although the machines in those experiments technically perform macroscopic work, they fall far short of what researchers think is possible. To be useful, a molecular machine must produce a continuous net output: repeating a task without first undoing all the work it’s already done. That will require not just a two-state switch that can toggle back and forth, but a complicated assembly of interconnected molecular parts that can attach and detach. Earlier this year Leigh and colleagues demonstrated a simple version of how such a machine might work: They built a molecular crane that picks up a small cargo molecule and transports it 2 nm along a rigid molecular platform. 8
What if, instead of picking up the cargo and putting it back down, two cranes were to pick up a molecule and pull it apart in a controlled way? “I think the first truly useful molecular machines will be ones that perform molecular tasks rather than interfacing with macroscopic objects,” says Astumian. “If we could create or break chemical bonds by using external energy to manipulate the positions of reactants rather than by relying on thermal energy, it could provide a route to the synthesis of high-energy compounds that would otherwise be impossible to make in good yield.”
Significantly, Leigh’s entire crane apparatus was all one piece: The crane was connected to the platform by a bond that could be controllably swiveled, and at every step in the process, the cargo was chemically bonded to the crane, the platform, or both. If the parts had disconnected at any stage, they would have been uncontrollably swept apart by Brownian motion of the solvent. Says Katsonis, “A big outstanding challenge is to find ways to decouple molecular machines from the Brownian storm—and further, to couple them to a functional environment.”
For simple systems, that coupling can be achieved by attaching molecules to a flat surface or molecular platform. For more complicated machines with more than a few moving parts, it may require a scaffold of polymers analogous to the network of microtubules that make up the cytoskeleton. “Kinesin, a motor protein in cells, functions by walking progressively down a microtubule, not by detaching and rebinding in a different place,” explains Anne-Sophie Duwez of the University of Liège in Belgium. “Coupling artificial molecular machines to a polymer scaffold could be a way to exploit submolecular motions to perform useful tasks on the single-molecule, micro-, or macroscopic level.” Says Chris Jarzynski of the University of Maryland, “We know it’s not a pipe dream to imagine these complex tasks, because biomolecules prove it’s possible.”
Even without such complex assemblies, though, many questions of fundamental interest arise from the controllable degrees of freedom of mechanically interlocked molecules and overcrowded alkenes. For example, Edie Sevick and colleagues at the Australian National University have theoretically investigated the behavior of what they call piston rotaxanes: several rings threaded onto a single axle that behave almost like a one-dimensional gas. They’ve also analyzed the daisy-chain molecule of figure 1c as a switchable liquid crystal. 9 “There’s so much more to be done—even little things,” says Sevick, “and there’s so much potential for collaboration between physicists and chemists.”
The laureates
Jean-Pierre Sauvage was born in 1944 in Paris. In 1971 he earned his PhD from Louis Pasteur University, now part of the University of Strasbourg. He was the first doctoral student of Jean-Marie Lehn, who went on to win the Nobel Prize in Chemistry in 1987. After a year as a postdoc at the University of Oxford and several more as a researcher in Lehn’s group, Sauvage established his own lab in Strasbourg in 1980. He is now an emeritus professor.

Sauvage
SCHRODER/UNISTRA

J. Fraser Stoddart was born in 1942 in Edinburgh, Scotland, and obtained his PhD in 1966 from Edinburgh University. He has held faculty positions at Sheffield University, Birmingham University, UCLA, and Northwestern University, where he remains today. In November 2016 he released a book, The Nature of the Mechanical Bond: From Molecules to Machines (Wiley), cowritten with his former student Carson Bruns.

Stoddart
NORTHWESTERN UNIVERSITY

Ben Feringa was born in 1951 in Barger-Compascuum, a small town in the northeast corner of the Netherlands. He earned his PhD in 1978 from the University of Groningen. Other than six years as a research scientist for Royal Dutch Shell, he has spent his career at Groningen, where he became a professor of organic chemistry in 1988.

Feringa
UNIVERSITY OF GRONINGEN

References
1. C. O. Dietrich-Buchecker, J. P. Sauvage, J. P. Kintzinger, Tetrahedron Lett. 24, 5095 (1983). https://doi.org/10.1016/S0040-4039(00)94050-4
2. R. A. Bissell et al., Nature 369, 133 (1994). https://doi.org/10.1038/369133a0
3. M. C. Jiménez, C. Dietrich-Bucheker, J.-P. Sauvage, Angew. Chem. Int. Ed. 39, 3284 (2000). https://doi.org/10.1002/1521-3773(20000915)39:18<3284::AID-ANIE3284>3.0.CO;2-7
4. N. Koumura et al., Nature 401, 152 (1999). https://doi.org/10.1038/43646
5. J. Berná et al., Nat. Mater. 4, 704 (2005). https://doi.org/10.1038/nmat1455
6. R. Eelkema et al., Nature 440, 163 (2006). https://doi.org/10.1038/440163a
7. S. Iamsaard et al., Nat. Chem. 6, 229 (2014). https://doi.org/10.1038/nchem.1859
8. S. Kassem et al., Nat. Chem. 8, 138 (2016). https://doi.org/10.1038/nchem.2410
9. H. He, E. M. Sevick, D. R. M. Williams, J. Chem. Phys. 144, 124901 (2016).https://doi.org/10.1063/1.4943098
More about the authors
Johanna L. Miller, jmiller@aip.org





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}