A supercooled protein refolds unexpectedly
DOI: 10.1063/PT.3.4383
Human bodies have a narrow range of temperatures at which they function properly. Proteins behave similarly: At ambient temperatures they fold as needed for their biological purposes, but if they get too hot or too cold, their structures unravel. The details of what happens to proteins away from their conformational sweet spot and how or why they denature could provide insight into how they manage to find their functional forms at physiological conditions in the first place.
Proteins found in nature don’t exist in a vacuum, and the molecules surrounding them affect their behavior (see the article by Diego Krapf and Ralf Metzler, Physics Today, September 2019, page 48 ). It’s therefore not enough to consider only how interactions within a protein change with temperature; to fully understand a protein’s behavior, the dynamics and structure of the solvent—typically water—must also be taken into account.
At the intersection of protein folding and water’s molecular dynamics Daniel Kozuch, Frank Stillinger, and Pablo Debenedetti of Princeton University noticed something unexpected in their simulations. 1 Earlier work 2 led by Debenedetti had investigated the cold denaturation of Trp-cage, a 20-amino-acid model protein, in liquid water supercooled to 210 K. Those simulations, as expected, had shown a peak in the fraction of folded proteins at room temperature followed by a steep drop-off as the temperature decreased. But when the researchers lowered the temperature even further in their latest study, they found a surprising result: At 194 K, the proteins refolded. 1
The well-tempered ensemble
Experimental studies of protein folding are challenging because of the short length and time scales on which the folding occurs (see Physics Today, October 2019, page 21 ). Molecular dynamics simulations and theory—the tools employed by Debenedetti’s group—are therefore indispensable because they can provide otherwise inaccessible details.
Fast folding may be an impediment for experiments, but it’s a boon for simulations because it makes them more time efficient and less computationally expensive. Trp-cage, illustrated in figure
Figure 1.

Ambient and low-temperature structures for the protein Trp-cage reflect its cold denaturation. The α-helix (purple) remains stable, but the 310-helix (blue) seen at ambient temperatures unfolds at 224 K. A tryptophan amino acid (red) sits in the protein’s core. (Adapted from ref.

Molecular dynamics simulations mimic the stochastic motion of a physical protein; they evolve a protein from an initial to a final configuration by navigating through the protein’s free-energy landscape and finding a global minimum. Each computational step represents a small, random physical fluctuation in the protein’s conformation that happens on the femtosecond time scale. But a protein’s free-energy landscape is vast and complex. If the simulated protein randomly explored that entire space in femtosecond steps, it would take an impractically long time to find its final state.
To bridge the gap between experimental and simulation time scales, computational scientists use enhanced sampling methods that guide the protein’s steps through the free-energy landscape to help it explore more efficiently. 3 In their 2016 paper on simulating Trp-cage, Debenedetti and his group used parallel tempering—a technique originally developed to deal with slow dynamics in simulations of low-temperature spin glasses—to sample the protein’s conformational states. Also known as replica exchange molecular dynamics, parallel tempering helps the evolving protein access more states by running multiple copies of the simulation simultaneously and by periodically exchanging configurations at different temperatures. Basically, it helps each copy avoid getting stuck.
Parallel tempering enabled the researchers to simulate the protein at temperatures down to 210 K. But, says Debenedetti, “at low enough temperatures it was just impossible to equilibrate the system in reasonable times.” Thermal fluctuations had just gotten too small. He and his collaborators therefore turned to an enhanced version of parallel tempering that employs the well-tempered ensemble. 4 The updated technique reweights configurations to help the simulated protein overcome large free-energy barriers in fewer, more efficient steps.
Protein variations
Debenedetti’s previous study of the cold denaturation of Trp-cage showed the unfolding of the small helix (blue) shown in figure
Proteins are known to denature at low temperatures, so that result wasn’t a surprise; the researchers were focused on delineating the protein’s low-temperature thermodynamic properties, such as the free energy of unfolding and the heat capacity. But when Kozuch and coworkers looked at the fraction of folded proteins at even lower temperatures, things unexpectedly changed. The cold denatured configuration from the previous simulations appeared again, but at the lowest temperatures the folded fraction quickly increased, from around 10% at 200 K to nearly 100% at 180 K, as shown in figure
Figure 2.

Low-temperature refolding of simulated Trp-cage occurs below 200 K as the surrounding water molecules become increasingly tetrahedrally coordinated. Above that temperature, the simulated protein behaves as expected: The fraction of proteins in folded states peaks at ambient temperature decreases as the protein gets too hot or cold. (Adapted from ref.
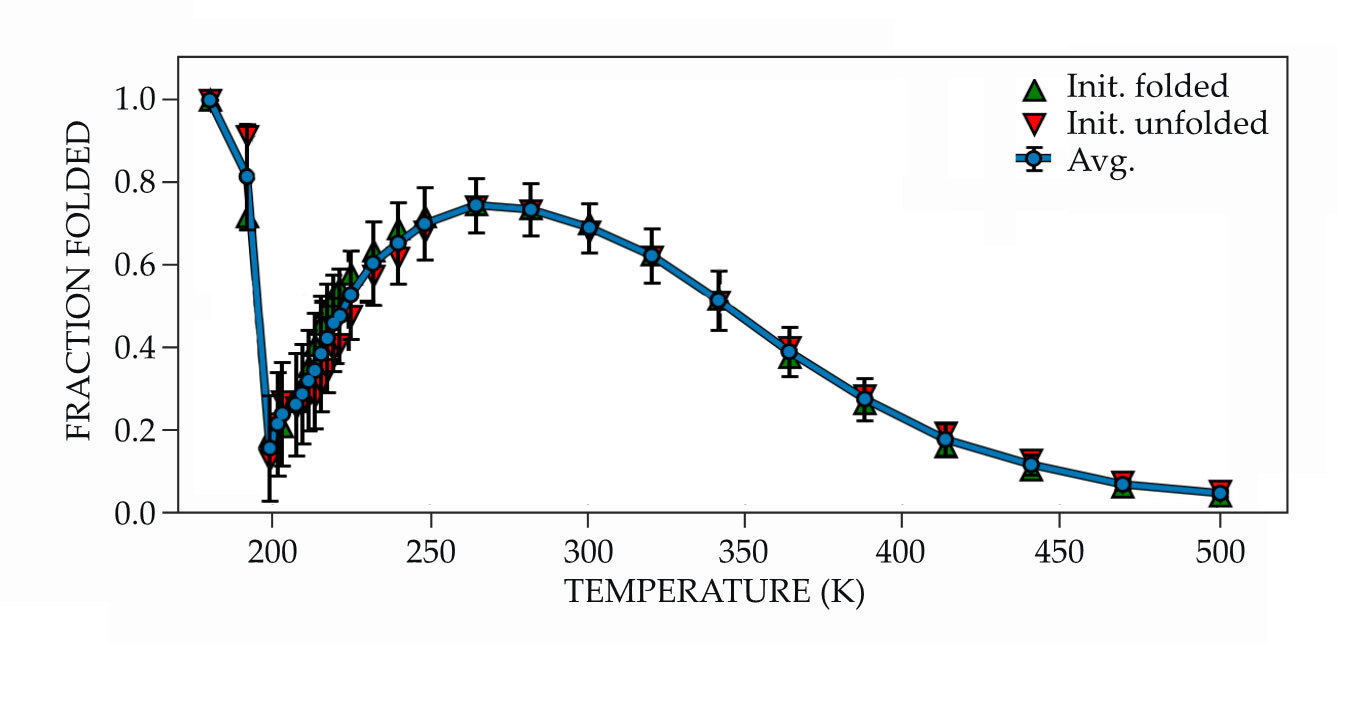
The results of Kozuch’s simulations were initially met with skepticism. “I really pushed back,” says Debenedetti. “This study took a long time. I had Daniel repeat the calculations many times.” But the results were robust. That the protein arrived at the same state regardless of whether it began folded or unfolded confirmed that the result reflected the underlying energy landscape and was not just an artifact.
The supercooled folded structure was remarkably similar, though not identical, to that at ambient temperature. Water molecules hydrated the folded protein’s core at room temperature, whereas the supercooled structure had a more compact hydrophobic core.
To seek an explanation for that structural difference, the researchers turned to the surrounding water molecules. Unlike most liquids whose densities increase as they get colder, water reaches its maximum density at 4 °C and then becomes less dense with cooling. The formation of short-lived hydrogen bonds at low temperatures generates transient connections between molecules, thereby increasing the average volume per molecule. By the time water reaches its minimum density, nearly all of the molecules are tetrahedrally coordinated. In the simulations, water’s minimum density and the protein refolding occurred synchronously at 195 K. (For more on the unusual behavior of supercooled water, see the article by Pablo Debenedetti and Gene Stanley, Physics Today, June 2003, page 40 .)
It’s no accident that the protein’s cold refolding coincided with water’s evolution to that low-density state. The researchers attribute the compact core’s formation to water’s increased order. Although the simulated water remained liquid and had no long-range order, on short length and time scales, the molecules were tetrahedrally coordinated. Solvating the protein’s core would have disrupted that order, so instead the water was expelled; hence the core’s collapse. Water’s role in reforming the helix is less clear, but it’s likely a factor. “Biology happens in water,” points out Debenedetti. “I would be really surprised if water played no role.”
Aqueous oratorio
Accurately capturing water’s low-temperature dynamics is a challenge. Many computational models for water exist, and although none are perfect, the TIP4P/2005 model used by Kozuch and coworkers is considered one of the best among classical models. It still has its shortcomings; for example, it places the water’s ambient-pressure melting temperature at 252.1 K, more than 20 K below its actual value. That means the researchers’ simulations at 200 K are actually only 52 K below freezing, not 73 K. But, importantly, the model has been shown to capture much of water’s known behavior—particularly its anomalous dynamics far from ambient conditions—and its complex crystalline phase diagram.
The researchers knew water could influence the protein’s behavior, which is why they wanted to capture its dynamics as accurately as possible. In fact, Debenedetti originally wanted to study how Trp-cage’s behavior would change around a liquid–liquid phase transition that has been seen in previous simulations of water. 5 But simulating the protein at the low temperature and high pressure necessary to reach that transition was unexpectedly difficult because the system took an extraordinarily long time to equilibrate. Luckily for the researchers, decreasing only the temperature was enough to uncover unexpected and intriguing behavior.
Although proteins don’t run the risk of becoming supercooled in vivo, the simulated temperatures and cooling rates are physically relevant for preparing cryo-electron microscopy and cryo-preservation samples. Now that they know where to look, the researchers are repeating their calculations on other proteins to see whether the refolding effect is more general.
References
1. D. J. Kozuch, F. H. Stillinger, P. G. Debenedetti, J. Chem. Phys. 151, 185101 (2019). https://doi.org/10.1063/1.5128211
2. S. B. Kim, J. C. Palmer, P. G. Debenedetti, Proc. Natl. Acad. Sci. USA 113, 8991 (2016). https://doi.org/10.1073/pnas.1607500113
3. Y. I. Yang et al., J. Chem. Phys. 151, 070902 (2019). https://doi.org/10.1063/1.5109531
4. M. Bonomi, M. Parrinello, Phys. Rev. Lett. 104, 190601 (2010). https://doi.org/10.1103/PhysRevLett.104.190601
5. J. C. Palmer et al., Nature 510, 385 (2014). https://doi.org/10.1038/nature13405





{kind=link}
{kind=link}