Strange interfacial molecular dynamics
DOI: 10.1063/PT.3.4294


Biological functions such as gene regulation and metabolism in living cells rely on highly specific molecular interactions. The structure and dynamics of interfaces—from the nanoscale surfaces of intramolecular domains and the molecular surfaces of proteins, to the mesoscale surfaces of organelles, and even to the microscale surfaces of live cells—mediate those interactions. Our understanding of how interfaces evolve and how they couple to their complex environments is still developing, and several Nobel Prize–winning technologies have aided the endeavor to understand them (see box
Box 1. Molecular dynamics and the Nobel Prize
Four Nobel Prizes have been awarded for work directly relevant to our current understanding of the atomistic nature of matter and molecular dynamics. Jean Perrin received the 1926 Nobel Prize in Physics for his work on the diffusion of microscopic particles and colloidal sedimentation and for introducing systematic single-particle tracking. The 2013 Nobel Prize in Chemistry was awarded to Martin Karplus, Michael Levitt, and Arieh Warshel for their contributions to computational multiscale models, which paved the way for the molecular dynamics simulations presented here. In 2014 the Nobel Prize in Chemistry was awarded to Eric Betzig, Stefan Hell, and William Moerner for their work on superresolution microscopy, a technique that has allowed researchers to track individual molecules in live biological cells. And for the 2018 Nobel Prize in Physics, half went to Arthur Ashkin, and the other half to Gérard Mourou and Donna Strickland, for their advances in laser physics and optical tweezers, which paved the way for groundbreaking research in probing biological systems by exerting piconewton forces on single molecules.
Here we discuss three examples of interfacial molecular dynamics: the distance fluctuations between the interfaces of internal protein domains, the coupling of proteins and lipid membranes to their surroundings, and the dynamics within lipid membranes—the thin layer separating a biological cell or an organelle from its surroundings. Even though the protein and membrane systems are highly dissimilar, their dynamics share many common fingerprints. Experiments and simulations demonstrate that both systems display rich and counterintuitive dynamics with strong deviations from researchers’ expectations. Experimental evidence has shown that interfacial dynamics in biological systems involve memory effects, which in turn affect molecular biological function in ways that are still not well understood.
Figure 5.

Trajectories of sodium channels moving on an energy landscape within the surface of hippocampal neurons. (Based on data from E. J. Akin et al., Biophys. J. 111, 1235, 2016, doi:10.1016/j.bpj.2016.08.016 .)

Until a few years ago, biological interfaces were studied primarily in terms of their chemical compositions and structures. It is becoming increasingly clear, however, that interfacial dynamics also are critical to the function of living organisms. They mediate processes such as the folding and internal dynamics of proteins, the formation of molecular complexes that can require multiple collision events to trigger specific binding, and the highly selective passage of molecules across membranes. Molecules in the water layer around large proteins and membranes not only associate locally with charge groups on individual amino acids in proteins and lipid membrane molecules; they also affect longer-range stabilization by forming so-called molecular water bridges. Water molecules mediate interactions between protein domains and membrane lipids through those bridges and establish a transient network that contributes to the stability of protein and membrane architectures. Molecular water bridges thereby affect the fluctuations in the structures of proteins and membranes and, in turn, influence the efficiency of biomolecular reactions.
Single particle paths
Membranes and molecules at interfaces are continuously bombarded by other small molecules and larger complexes in the surrounding aqueous environment. Thus, one may expect the dynamics of interfacial molecules to be dominated by diffusion as described in the theoretical works of Albert Einstein
1
and Marian Smoluchowski.
2
Within their framework, molecules are always jittering around randomly, completely independent of their pasts. Even for simple uncorrelated Brownian motion in which the mean squared displacement (MSD) is given by
Box 2. Mandelbrot’s fractals
Benoit Mandelbrot connected the notion of fractals to the geometric complexity of natural patterns. 3 He noted that “clouds are not spheres, mountains are not cones, [and] coastlines are not circles.” Physicists are trained to study processes in terms of their specific time or length scales. In many natural patterns, however, the number of distinct scales involved is so large—more detail appears upon further magnification—that for all practical purposes, it is infinite. Such a system is called scale-free. Brownian trajectories share the scale-free property of fractal patterns. Notably, the surfaces of proteins can have a fractal topology, which must be carefully considered when examining motion within such a structure. Diffusion in a fractal is different from diffusion in a Euclidean space because the ramified, tortuous underlying structure pushes the random walk to retrace its path. Thus, the increments of diffusion within a fractal have negative autocorrelations. A step in one direction is likely followed by a step in the opposite direction, which is also seen in the diffusion of tracer particles in viscoelastic environments. An interesting feature of most fractals and of random walks is their self-similarity. If the path is cut into smaller pieces, each piece appears to be statistically the same as the whole.
For hundreds of years, physicists have been studying the motion of individual particles to understand the physics of large systems. Their basic premise is that observed macroscopic properties arise at the most fundamental level from the dynamics and interactions of the individual components. To make sense of matter’s behavior, one should probe the physics of its constituent parts—atoms and molecules. Single-particle tracking has a long tradition that originated with Jean Perrin’s careful protocols, and what appeared to be science fiction not long ago is now routinely practiced: Careful experiments based on fluorescent tagging allow researchers to follow the motion of individual molecules in living biological cells. 4 Today, researchers can measure the locations of individual molecules with nanometer precision while following their motion with millisecond temporal resolution. The gap between the accessible time scales of simulations and experiments is continuously narrowing, and a significant overlap between simulated and experimentally measured dynamics is already possible.
Protein reconfiguration
Proteins—polymers composed of amino acid monomers with specific monomer–monomer interactions—are responsible for most of the essential processes in a living cell, including signaling, active transport, cellular metabolism, and modification of other proteins. A protein’s transition from one conformation to another takes place in the heterogeneous energy landscape of the protein’s highly dimensional phase space.
To function, most proteins in solution first fold into a predetermined structure. Once folded, they typically undergo major conformational changes. Transitions between different configurations are dominated by many small, local conformational changes that add up to a larger protein reconfiguration. The protein crosses a hierarchy of energy barriers but also encounters geometric hindrance, which is often neglected in models. A stark indication of the complexity of protein dynamics was shown in experiments by Hans Frauenfelder at the University of Illinois at Urbana-Champaign in the 1970s. They revealed that the distribution of binding times for ligands showed a power-law scaling over several decades in time. However, at the time it was not possible to tell whether the dynamics were a heterogeneous ensemble effect or due to an individual molecule.
In 2003 Sunney Xie’s group at Harvard University used fluorescent imaging methods in single-molecule experiments; their work showed that internal conformation fluctuations take place on a strikingly large range of time scales, from hundreds of microseconds to seconds. The picture that emerged is one of conformational states with a broad power-law distribution of trapping times, or fractal time. Because of that distribution of trapping times, the distance
The single-molecule experiments were recently put into a broader perspective in a supercomputing study. Jeremy Smith’s group at Oak Ridge National Laboratory confirmed that
Figure 1.

The structure of a protein and its surrounding water molecules exhibit intriguing dynamics. (a) The yeast globular protein phosphoglycerate kinase has three domains, shown here in red, yellow, and blue. Its internal fluctuations can be characterized by the relative position

A paradigm shift is now under way in understanding protein dynamics, and the effects of the interfacial fluctuations can no longer be neglected. Those effects are likely at the core of protein conformational changes and function. To gain mechanistic insight into the emergence of complex organization in proteins, it is essential to probe their dynamics at the protein–water interface, which can dramatically alter intramolecular interactions.
Liang Hong’s group at Shanghai Jiao Tong University provided one piece of evidence for strange dynamics at the protein–water interface through neutron scattering experiments of proteins hydrated by approximately a single water layer. The experiments were performed in tandem with molecular dynamics (MD) simulations.
7
The researchers demonstrated that the water molecules on the surfaces of two proteins—cytochrome P450 and green fluorescent protein—exhibit subdiffusion: They both had MSDs of the form
The MD simulations showed a gradual change from subdiffusion to nearly normal diffusion
What would cause the fractal immobilization times observed for internal and external protein interface dynamics? In the 1970s Harvey Scher at Xerox and Elliott Montroll at the University of Rochester showed that such scale-free dynamics may readily emerge from energetic traps with random depths (see box
Box 3. Fractal-time Pearson walks
When Karl Pearson conceived the random walk, he envisioned a man walking a given distance in one direction, turning in a random direction, and repeating the process. In the continuum limit, the process describes Brownian diffusion. Now consider the more general case when the random walker can rest for random times between sojourns. That could describe the motion of a water molecule that spends time in pockets on the surface of a protein.
The theory of such a stochastic process was introduced in 1965 by Elliott Montroll and George Weiss, then at the Institute for Defense Analyses and the National Institutes of Health, respectively. Their work gave rise to the famed continuous-time random walk (CTRW), a term coined by Harvey Scher in 1973. Scher and his colleague at Xerox, Melvin Lax, brilliantly succeeded in modeling their unconventional observations of electrical conduction in amorphous semiconductors in terms of a CTRW with scale-free rest times. Because the distribution of times was similar to Mandelbrot’s geometric fractals, Mike Shlesinger from the Naval Research Office called it a time fractal.
Such dynamics occur when the distribution of rest times
Probing single molecules in their natural habitat is crucial to understanding their dynamics and function. When solvated in water, proteins perform extravagantly choreographed ballet dances that often include major displacements of subunits around hinge-like bonds. In addition to that motion, the liquid inside a cell is crowded with large, squishy biomolecules that are incessantly colliding like bumper cars. Over time scales of collisions between the large crowding molecules in the cellular environment, proteins trapped in certain conformations or surface water molecules immobilized for long times might be unlocked by nearby colliding molecules, which would lead to a renewal of the motion: The molecules could resume their vivid choreography and restart the ageing process. That picture is still speculative, but we expect the internal protein dynamics and the motion of the surface water to still be anomalous at least below typical renewal times.
In and around membranes
The same interfacial dynamics that affect single proteins are also important for lipid bilayer membranes that form the surfaces of cells and organelles. First consider the simplest model system for a biological membrane: a self-assembled bilayer of lipid molecules of identical chemistry (figure
Figure 2.

Snapshots of lipid bilayer membranes with adjacent water layers from molecular dynamics simulations show the effects of membrane disorder. (a) A single-component lipid bilayer (green) exists in a disordered liquid phase. Water molecules (red and white) surround the bilayer. (b) When cholesterols (orange) are added, the bilayer transitions to a liquid ordered phase. Note that the bilayer width increases with decreasing lipid-tail entropy. (Courtesy of Matti Javanainen.)
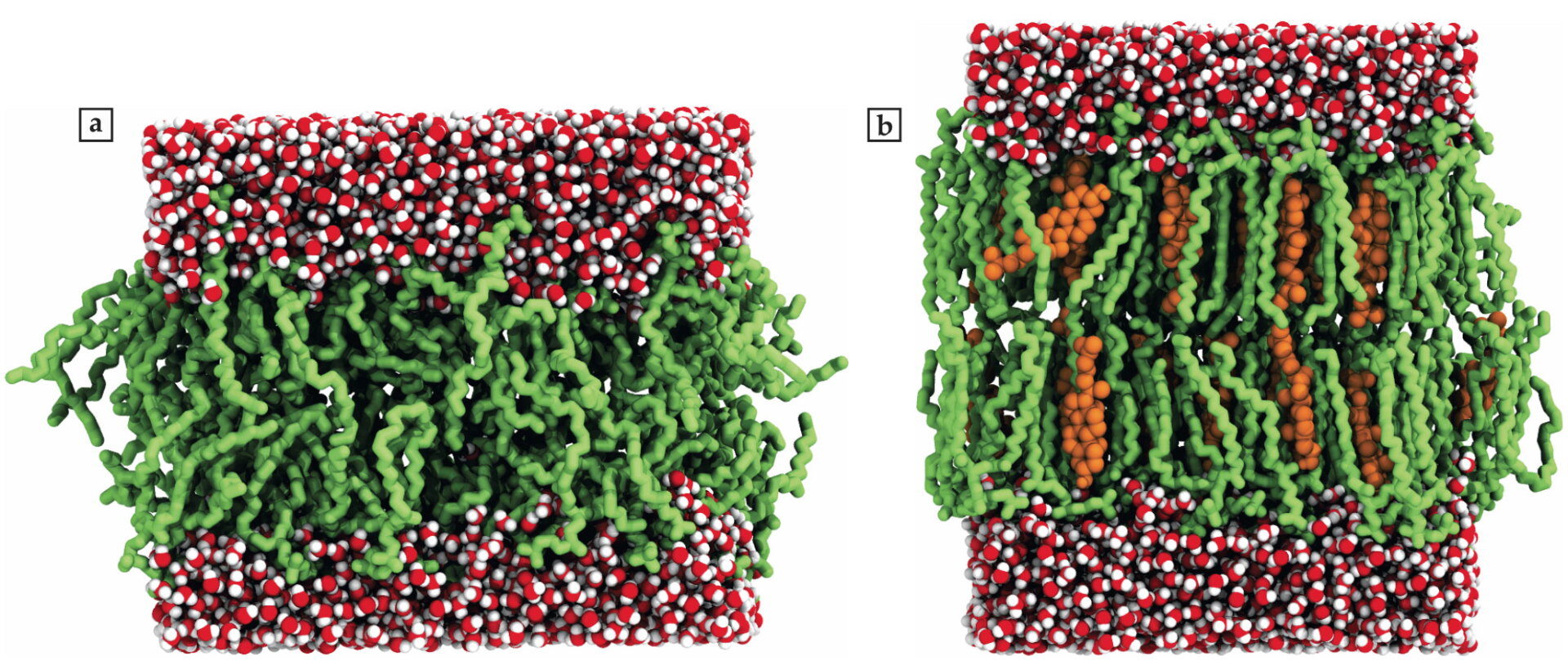
When cholesterol molecules—a type of lipid essential for the structural identity of animal cell membranes but better known for causing cardiovascular diseases—are added to the bilayer, they settle snugly between the tail groups of the bilayer lipids. As shown by Ilpo Vattulainen’s group at the University of Helsinki, cholesterols decrease the accessible degrees of freedom of the membrane lipids. That restriction causes a more ordered bilayer structure and, because the lipid tails become more elongated, an increased membrane width (figure
The correlation between higher membrane disorder and longer-lasting anomalous diffusion is corroborated when proteins are added to the bilayer. Lipid bilayers in biological membranes comprise various lipids with different chemistries, and they are studded with membrane proteins much larger than the surrounding lipids (figure
Figure 3.

The membranes of live cells are heavily crowded by embedded proteins. In order for coarse-grained molecular dynamics simulations to be realistic, a lipid bilayer is decorated with multiple membrane-embedded proteins.

The lipids and proteins in bilayers interact both with each other and with their surroundings. To see the effects of those interactions, Eiji Yamamoto and colleagues at Keio University in Tokyo studied the motion of water molecules and proteins at the water–membrane interface. Their all-atom MD simulations showed that water molecules within 3.5 Å of the lipids are subdiffusive with
Going one step further, Yamamoto and others have studied the surface motion of a protein that specifically binds to lipid head groups in multiscale MD simulations.
12
The results again reveal anomalous heterogeneities in the protein’s diffusion with intermittent localization patterns characterized by an exponent
How do those observations translate into observations of membrane dynamics in complex living cells? Different experimental studies reveal two crucial factors. Due to the increased complexity of the study cases described above, subdiffusion of membrane proteins may reach longer macroscopic time scales of hundreds of seconds, and their motion can be strongly influenced by interactions with the intracellular scaffolds of semiflexible polymers that support the membrane. Scale-free trapping times cause membrane proteins to exhibit ageing dynamics on similarly long time scales. 13
Using a combination of single-particle tracking and superresolution imaging in live cells, researchers found that the actin cytoskeleton that lies underneath the membrane forms a scale-free landscape, shown in figure
Figure 4.

The cytoskeleton structure vicinal to the membrane of a human embryonic kidney cell can be probed by superresolution microscopy. The cortical cytoskeleton forms a scale-free fractal network (orange). The green lines show the compartmentalization of the membrane that is induced by the fractal actin structure. (Adapted from ref.

Origins and relevance of anomalous dynamics
The internal dynamics of proteins and membranes and the dynamics of surface-associated molecules are very rich and dominated by anomalous diffusion on many time scales. The physical nature of the subdiffusive dance has two faces. One is a random-walk-like, intermittent motion with scale-free immobilization events, possibly with superimposed local small-amplitude jitters. Such processes with diverging mean waiting time are inherently non-Gaussian. The other face is a viscoelastic-type anomalous diffusion governed by anticorrelated but stationary dynamics. Such antipersistent motions with power-law step correlations are normally Gaussian, 7 but in a heterogenous medium they may also become non-Gaussian. 10
Ageing effects, identified on many time scales, cause a perpetual decrease of the effective diffusivity over time. In cases when ageing persists over many time scales, one must consider how much time has elapsed since the system was initialized. For a protein, that initialization might be when it was synthesized by a ribosome. Knowing when the dynamics began evolving is necessary to extract relevant physical parameters such as the value of the anomalous diffusion coefficient. We may ask whether ageing in the internal motion of individual proteins will persist over longer times in the crowded environment of living cells. Due to bombardment by other biopolymers, proteins may experience considerable impacts, a kind of massage causing the unlocking of stuck configurations and thus a rejuvenation of the dynamics that resets the molecule’s internal clock and causes ageing to start again.
Scientists are still collecting evidence from experiments and simulations in an effort to understand the physical and biochemical consequences of interfacial dynamics. At ultrashort and short time scales, the observed anomalous diffusion in complex systems may point to fundamental interactions such as caging effects either in the dense array of lipids 8 or of the surface water. 7 Considering that water bridges can connect surface amino acids in a protein or connect lipid molecules in a membrane, they may not only add to the stability of protein or membrane architectures but also slow their motion. Moreover, when two biopolymers come close in the crowded cytoplasm of the cell, such water bridges may generate longer mutual contact times and thus facilitate chemical reactions and oligomerization.
The viscoelastic component in several of the observed systems appears to be a generic behavior in soft-matter systems. Because of the antipersistence of their trajectories, individual particles may remain mobile but are likely to be pushed back to their previous position over long times. Returning to the same location may increase the likelihood of repeated reaction attempts and further be beneficial in forming complexes, especially in real, highly mixed membranes. Being slow may not always be a virtue, but within cells it may underlie function.
The observed strange interfacial molecular dynamics reflect both energetic and dynamic disorder. The journey into exploring those effects and their implications for biological function has likely just begun.
References
1. A. Einstein, Ann. Phys. 322, 549 (1905). https://doi.org/10.1002/andp.19053220806
2. M. Smoluchowski, Ann. Phys. (Leipzig) 21, 756 (1906). https://doi.org/10.1002/andp.19063261405
3. B. B. Mandelbrot, The Fractal Geometry of Nature, W. H. Freeman (1983).
4. K. Nørregaard et al., Chem. Rev. 117, 4342 (2017). https://doi.org/10.1021/acs.chemrev.6b00638
5. H. Yang et al., Science 302, 262 (2003). https://doi.org/10.1126/science.1086911
6. X. Hu et al., Nat. Phys. 12, 171 (2016). https://doi.org/10.1038/nphys3553
7. P. Tan et al., Phys. Rev. Lett. 120, 248101 (2018). https://doi.org/10.1103/PhysRevLett.120.248101
8. S. Gupta et al., J. Phys. Chem. Lett. 9, 2956 (2018). https://doi.org/10.1021/acs.jpclett.8b01008
9. J.-H. Jeon et al., Phys. Rev. Lett. 109, 188103 (2012). https://doi.org/10.1103/PhysRevLett.109.188103
10. J.-H. Jeon et al., Phys. Rev. X 6, 021006 (2016). https://doi.org/10.1103/PhysRevX.6.021006
11. Y. von Hansen, S. Gekle, R. R. Netz, Phys. Rev. Lett. 111, 118103 (2013). https://doi.org/10.1103/PhysRevLett.111.118103
12. E. Yamamoto et al., Sci. Rep. 5, 18245 (2016). https://doi.org/10.1038/srep18245
13. A. V. Weigel et al., Proc. Natl. Acad. Sci. USA 108, 6438 (2011). https://doi.org/10.1073/pnas.1016325108
14. S. Sadegh et al., Phys. Rev. X 7, 011031 (2017). https://doi.org/10.1103/PhysRevX.7.011031
15. G. de Wit et al., Biophys. J. 114, 2945 (2018). https://doi.org/10.1016/j.bpj.2018.05.007
16. J. Ehrig, E. P. Petrov, P. Schwille, Biophys. J. 100, 80 (2011). https://doi.org/10.1016/j.bpj.2010.11.002
17. C. Manzo et al., Phys. Rev. X 5, 011021 (2015). https://doi.org/10.1103/PhysRevX.5.011021
More about the authors
Diego Krapf is an associate professor of electrical and computer engineering at Colorado State University in Fort Collins. Ralf Metzler is a chair professor of theoretical physics at the University of Potsdam in Potsdam, Germany, and is currently a Humboldt Polish Honorary Research Scholar.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}