Recollision physics
DOI: 10.1063/1.3563818
In 1906 Ernest Rutherford discovered that α particles deflect as they pass through a mica film. That experiment, which helped Rutherford identify the atomic nucleus, was a dramatic demonstration that collisions between particles could tell us about the structure of matter. Now, a century later, high-energy collisions between subatomic particles have revealed the fundamental building blocks of our world—quarks, muons, and so on—and lower-energy collisions have been central to understanding and harnessing nuclear physics.
Just over 50 years after Rutherford’s experiment, the laser was demonstrated. Since then, optical physics, which deals with interactions between light and matter, has developed powerful methods for exciting, probing, and controlling matter and its dynamics. The precision of optical experiments has reached the point where some of the most fundamental questions of particle physics can be tested better optically than by collisions.
Although optical and collision physics are traditionally considered separate disciplines sharing little, if any, overlap, the emerging field of recollision physics unites the two. In a recollision, the oscillating field of a laser pulse causes an electron to accelerate away from an atom or molecule and then, upon reversal of the field, careen back into its parent ion. Whereas traditional collision physics relies on large accelerators and magnets to arrange the collision, in recollision physics it is the laser field that provides the acceleration and the atom itself that provides the electron with which it is probed. Through recollision, optics gains access to the well-developed capability to probe the structure of matter via collisions (the focus of this article) and collision physics gains access to the capability to excite, probe, and control matter with light.
Two coherence transfers
Consider an atom illuminated by a pulse of coherent IR light. If the light is intense enough, roughly 1013 W/cm2 or higher (see box

Figure 1. The basics of recollision. In the first step, an intense pulse of coherent IR light skews the potential well (black) of an atom or molecule’s electron. That allows the bound-state electron wavefunction (blue, sketched as a Gaussian) to tunnel and split, creating a wavepacket in the continuum. There, the wavepacket is driven by the oscillating laser field in a semiclassical trajectory (red): It first accelerates away from the atom and then, upon reversal of the field, accelerates back toward its origin, recolliding with its parent ion. The process is conceptually similar to an optical interferometer. The relative wavelengths of the tunneling wavepacket and bound-state wavefunction are illustrated approximately to scale.
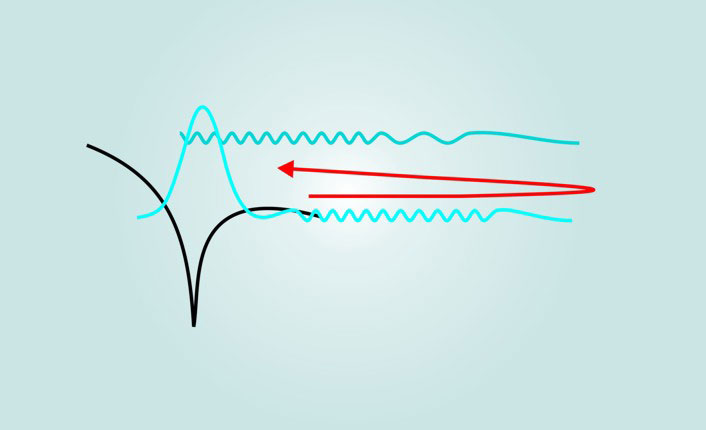
Once the electron has tunneled, the resulting wavepacket—now in the continuum, freed from the pull of its parent ion—is driven in a semiclassical motion by the laser field. The classical approximation of subcycle electron motion has a long history in plasma physics. 2 It is useful when many photons are involved.
What happens next depends on the polarization of the light pulse. If the polarization is circular, then as soon as any portion of the wavepacket emerges from the atom or molecule, it gets pulled by the field in constantly changing directions—first away from the ion, then laterally, and so on. The cusplike motion ensures that the wavepacket never returns to the ion of its birth.
If the beam is linearly polarized, different parts of the wavepacket will follow different trajectories, depending on when during the laser cycle they were born. The first parts of the tunneling-electron wavepacket to emerge from the atom experience a rising laser field—they accelerate away from the ion for more than one-quarter period, gaining a large momentum before the field reverses. That momentum causes them to drift away from the atom for more than an additional quarter period before reversing direction. Thus the momentum imparted by the reversed field falls shy of what’s needed to return those parts of the wavepacket to the parent ion; they oscillate and drift away, never to return. In contrast, those portions that tunnel after the field crests accelerate away from the ion for less than one-quarter period and then drift back to recollide with the ion within one laser period.
The recolliding electron wavepacket can be thought of as recombining with the bound-state wavefunction from which it tunneled. In the process, it radiates light with energy roughly equal to the sum of the wavepacket energy and the ionization potential and with a phase determined by the phase of the recolliding wavepacket and the structure of the bound-state wavefunction.
Tunneling transfers information about the bound-state phase to the wavepacket, the first coherence transfer of recollision physics; recombination transfers information about the recollision electron phase to the radiated light, the second. Typically, that radiation ranges from the UV to the extreme UV (XUV): The frequency increases, or blue-chirps, during the first half period after tunneling, then decreases, or red-chirps, during the next quarter period. If recollisions are driven repetitively by multicycle IR pulses, they create high harmonics of the driving frequency. If the recollision is controlled so that it occurs during only one half-cycle, a single attosecond XUV pulse is created. Experimentally it is even possible to select primarily the blue-chirped radiation of a single recollision.
Both high harmonic and attosecond pulse generation are important technical developments with major scientific implications. See, for example, the article by Henry Kapteyn, Margaret Murnane, and Ivan Christov (PHYSICS TODAY, March 2005, page 39 ) and other influential reviews given in reference . The present article, however, focuses on another aspect of recollision—the ability to gain information about atoms, molecules, and solids through ionization and recollision.
A laser STM?
The first step in the recollision process, laser tunneling, may itself provide valuable information about atomic and molecular structure. 4 In scanning tunneling microscopes (STMs), for example, the tunneling between a sharp, biased tip and a nearby surface serves as a spatial filter that is precise enough to resolve individual surface atoms. Can laser tunneling also serve as a spatial filter?
Atomic theory 5 states that a tunneling electron creates a wavepacket Ψc in the continuum characterized by its lateral momentum p ⊥—the momentum perpendicular to the laser’s electric field:

(1)
Here, IP is the ionization potential, E is the field at which the electron tunnels, and Ψb is the bound-state wavefunction. The basic structure of equation 1 can be understood qualitatively. The portion of the bound-state momentum wavefunction that is parallel to the laser polarization will ride higher on the barrier than those portions with some of their momentum in the lateral direction. Having less of the barrier left to penetrate, those low-lateral-momentum electrons will be the ones that populate the lion’s share of the continuum wavepacket. Thus tunneling in atoms is a directional momentum-space filter. If that is also true for molecules, then rotating the laser field around a molecule’s axis, for example, is analogous to translating an STM tip across a surface: The probability of ionization will depend on the orientation of the field relative to the molecular axis, just as the current in an STM depends on the position of the tip relative to the surface atoms.
In a conventional STM, an electron that is extracted from a surface disappears into the tip. The resulting current is what provides information about the surface structure. In a laser STM, an avalanche detector—such as a microchannel plate—can count the total number of electrons or ions created per laser shot at each angle between the molecular axis and the laser electric field. That count is the direct analogue of the current.
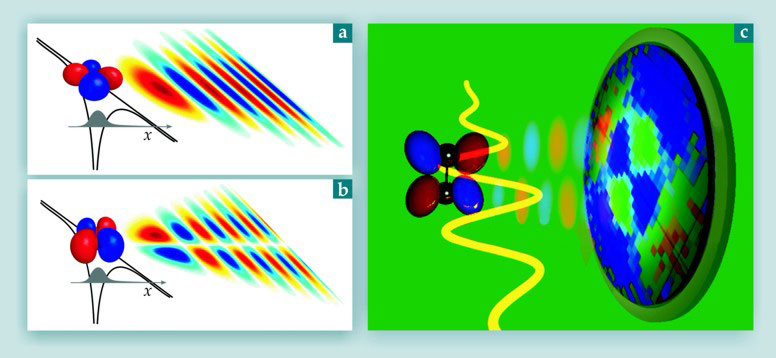
However, in a low-density gas even more information is available. It follows from equation 1 that the bound-state orbital leaves a deep imprint on the tunneling-electron wavepacket, not only through the angle dependence of the ionization probability but also through the angle dependence of the wavepacket structure. That imprint is illustrated in figure 2, panels a and b, which contrast wavepackets generated for two different orientations of the light polarization relative to the axis of an oxygen molecule. A two- or three-dimensional imaging detector like that illustrated in figure 2c can directly measure the transverse component of the electron wavepacket. As long as the electron does not recollide, nothing stands between it and the detector.

Figure 2. Recollision as an STM. (a) A laser field polarized in the x-direction creates a tunneling electron wavepacket peaked along the x-axis for an oxygen molecule aligned at approximately 45°. (The black line shows a snapshot of the laser field; the curve underneath shows the resulting distortion of the electron potential well. For the escaping wavepacket, red and blue denote the relative phase; the color intensity denotes the relative amplitude.) (b) If the molecule is aligned at 0° or 90°, the peak is replaced by a minimum. (c) With an appropriately placed detector (illustrated at right) and a fixed molecular alignment, the wavepacket can be imaged in the yz-plane. The illustration depicts actual experimental data; the alternating blue and green quadrants reflect the nodal structure of a wavepacket obtained for O2 molecules aligned perpendicular to the laser field. The red signal near the perimeter of the detector corresponds to recollision electrons that were elastically scattered from the parent ion. (Adapted from ref.
If initiated by 800-nm-wavelength light, tunneling effectively takes place over about 300 attoseconds during each half cycle. Even using 1.8-µm light, tunneling is confined to less than 1 fs. Since both 800-nm and 1.8-µm pulses can be reduced to a single cycle, tunneling can be confined to a half cycle. With such fine time resolution, it should be possible to use laser tunneling (or laser-induced electron diffraction, 6 when the electron recollides) to time-resolve intra-atomic electron dynamics and to follow structural changes during chemical transitions.
Given that the theory of multiphoton ionization of atoms and solids arose together, 1 one might suspect that the tunneling probability in a crystalline solid also depends on the angle between the polarization direction and the crystal axis. We have confirmed experimentally that it does. 7
Recollision as interferometry
Classical physics shows that portions of the tunneled electron wavepacket that recollide with its parent ion have a maximum energy

(2)
where U c is known as the ponderomotive energy—the electron’s energy of oscillation—q is the electronic charge, E 0 is the laser’s peak field during the cycle of ionization, m is the electronic mass, and ω is the laser angular frequency. (Although it may seem that the maximum energy of recollision does not depend on the atom or molecule, it really does. The atom or molecule selects the range of fields E 0 over which the electron can tunnel.)
For noble gas atoms and closed-shell molecules, typical recollision energies range from 1 to 103 eV. That corresponds to an electron wavelength between 10 and 0.3 Å, comparable to the interatomic separation in molecules and the distance between the nodes of bound-state wavefunctions.
It’s worth noting that the recollision truly is a collision and that 103 eV is by no means the energy limit. (As discussed in box

(3)
where S is the classical action of the electron wavepacket along its trajectory and IP is the ionization potential. In typical experiments, φ can reach 100 radians or more, depending on the trajectory and the light intensity and wavelength.
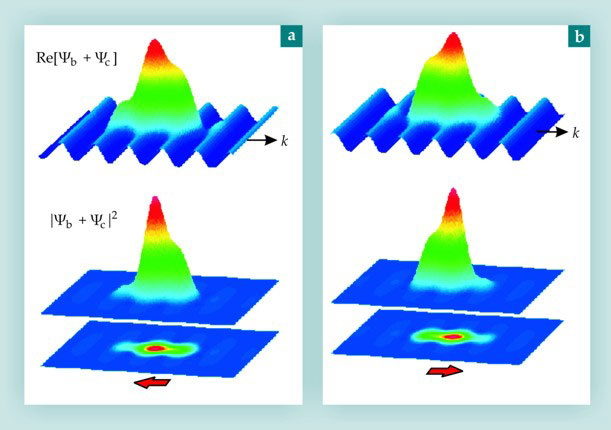
The superposition of the continuum electron wavepacket and the bound-state wavefunction creates an interference pattern like that illustrated in figure 3. As the recollision wavepacket moves, the interference pattern oscillates, creating an oscillating dipole that radiates UV and XUV light. Importantly, the phase of the emission is determined by the phase of the recollision electron and the structure of the orbital.

Figure 3. Generation of high harmonics via recollision. (a) The total wavefunction (top row), the total probability (middle row), and the two-dimensional projection of the probability function (bottom row) illustrate the dipole induced by the superposition of the approximately Gaussian bound-state wavefunction Ψb with the approximately sinusoidal recollision wavepacket Ψc. The red arrow indicates the orientation of the dipole. (b) The same superposition shown an instant later, after the recollision wavepacket has moved by half a cycle in the k direction, illustrates the dipole oscillation.
The recollision can be viewed, then, as having constructed an electron interferometer from a molecule’s own electrons. Tunneling—the first coherence transfer—serves as the beamsplitter. The same laser field that initiates tunneling also creates a coherent delay line for the electron, reminiscent of the delay line in an optical interferometer. The interferometer is read through the radiation emitted when the electron recombines with its bound state in the second coherence transfer. Our control over the electron interferometer rivals that of an optical interferometer: We can change the laser wavelength, intensity, or polarization or add a weak harmonic field to adjust the electron’s trajectory, just as a micrometer-controlled translation stage can be used to adjust the beam trajectory in an optical interferometer.
To the extent that a recollision can be thought of as a continuum electron recombining with the bound state to emit high harmonics or an attosecond pulse, it is closely related to single-photon photoionization—except that it happens in reverse. Tunneling can be viewed as creating an inverse photoelectron that recombines with the bound state. Thus measuring photons acts as a surrogate for measuring the photoelectron. The change in perspective is not just a formality. Rather, all of the power of optics to measure and exploit phase becomes available to inverse photoelectron spectroscopy.
Tomographic imaging of orbitals
In principle, optical interferometry can be used to decode just about everything that can be known about an optical pulse: The temporal structure of short pulses can be measured with interferometric techniques having such whimsical names as Spider, Rabbit, and Frog; spectra can be characterized with Fourier-transform interferometers; the spatial structure of beams can be determined with sheared interferometers. What is true for an optical pulse and an optical interferometer should also be true for an electron wavefunction and the recollision electron interferometer. Some might argue that wavefunctions cannot be measured—even in principle. Yet optics experiments suggest otherwise. Up to one unknown phase, both the intensity and field structure of an optical pulse are measurable.
My colleagues and I have posited a purely experimental method to measure an electron wavefunction. Assuming a single active electron and approximating the recollision electron as a plane wave in space—the strong-field approximation—the induced dipole can be written as

(4)
where Ψb is the bound-state wavefunction from which the electron tunneled, q is the electronic charge, and a[k(ω)] is the amplitude of the recolliding wavepacket.8 (Readers interested in multielectron systems should see reference .) The axes are chosen so that the recolliding electron wavepacket moves along the x-axis. Thus, by measuring the spectrum of the attosecond or high-harmonic emission, given by ω 4 d 2, we measure the product of two important quantities: the Fourier transform along the x-axis of the orbital multiplied by the vector r and the amplitude of the recollision electron. The latter is determined by the tunneling probability and the subsequent trajectory of the continuum wavepacket.
Equation 4 suggests a strategy to realize the general statement about the implications of interferometry with an electron interferometer. By measuring the single-molecule high-harmonic spectrum―its amplitude, polarization, and phase―and a[k(ω)], it’s possible to measure the 1D Fourier transform of the orbital. Taking different projections through the molecule and applying tomographic algorithms, one can then reconstruct the orbital. 10
To realize the tomographic procedure in an experiment, we must first be able to measure the single-molecule harmonic spectrum. One of the beauties of attosecond and high-harmonic beams is that a gas of atoms produces a phase-matched, macroscopic signal. For symmetric molecules at low to moderate gas densities, the phase-matched output is essentially an intense version of the single-molecule response, so that the macroscopic signal directly tells us about the physics at the level of a single atom or molecule.
Next we must be able to control molecular alignment to allow tomographic projections. To that end, laser techniques can be used to align an ensemble of molecules at any angle with respect to the polarization direction of the probe pulse (see box
From principle to practice
Although harmonic emission as a function of angle has been measured for numerous molecules, orbital images have been reported for only two cases: nitrogen and carbon dioxide. 10 Nitrogen is a particularly convenient test case. Because its highest occupied molecular orbital—the so-called Dyson orbital—is a σ orbital, the tunneling and recollision probabilities are roughly independent of the field polarization direction. One value of a[k(ω)] can serve for all angles.
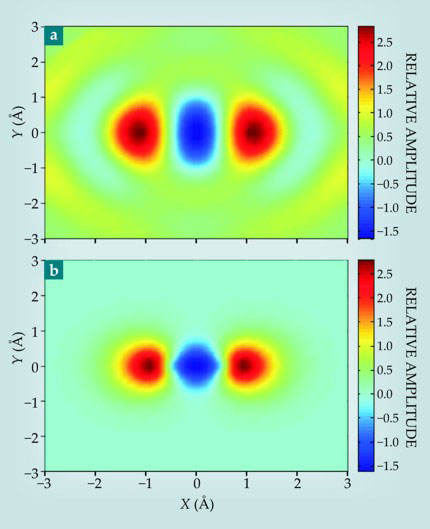
It is possible, then, to jointly determine a[k(ω)] and the spectral response of the experimental system, using an argon atom, which has similar ionization properties as N2, as a reference. We assume that the only orbital that ionizes in Ar is the 3p orbital pointing in the laser-field direction, and that it has the same orbital structure as the 3px orbital of a hydrogen atom. Other than that, with only the harmonic spectrum of Ar for reference, the measured angle-dependent harmonic spectra for N2, and a mathematical algorithm, we produced the orbital image shown in figure 4.

Figure 4. Recollision tomography. (a) The image of the highest occupied molecular orbital in a nitrogen molecule shown here was derived from a recollision experiment. The color indicates the relative amplitude. (b) The orbital image attained via the Hartree–Fock calculation is qualitatively similar. (Adapted from ref.
Notably, we were able to measure both the amplitude and relative phase of the wavefunction, just as we might measure the amplitude and relative phase of an optical pulse with sheared interferometry. Since the electron wavepacket is split from the initial wavefunction, both arms of the interferometer share a common, unmeasured phase.
It is important to emphasize that each of our assumptions—the recollision model, the strong-field approximation, and the 3px orbital structure of Ar—needs improvement. Nonetheless, our experimentally measured image of the Dyson orbital very much resembles those that have been calculated. The general principle—well established in optics—that interferometry allows full characterization of the waves seems transferable to electron orbital wavefunctions.
Why aren’t there experimental images of many other orbitals in the world’s scientific literature? In my view, it is largely because, as seen in equation 1, the tunneling rate and recollision probability in most molecules are highly angle dependent. That makes the selection of a reference atom difficult. In addition, at some molecular-frame angles, suppressed ionization and recollision in the highest occupied molecular orbital allows other orbitals to contribute to the signal. 9 In that regard, N2 was an especially fortunate choice; other molecules will require new approaches, some of which are already under development, 11 to avoid such problems.
Only scratching the surface
I have concentrated on laser pulse intensities that range from about 1013 to 1015 W/cm2. Since recollision requires only weak ionization, just a small extension beyond the intensities used in most previous ultrafast experiments has been necessary. The subtle shift, however, permits a completely new set of spectroscopies. Some—the laser STM, laser-induced electron diffraction, 6 , 7 and orbital tomography, 10 for instance—are qualitatively different from those that existed previously. Others simply extend ultrafast science into a new time and frequency range. In my opinion, the coherence transfers that underlie recollision physics will open a route to fully coherent high-harmonic spectroscopy. Powerful methods from conventional nonlinear optics, including transient-grating spectroscopy and heterodyne and homodyne detection, can now be deployed. 12
Recollision physics is so different from previous ultrafast methods that it can be applied on all time scales. Photochemistry, for example, unfolds on the femtosecond time scale. Therefore, many aspects of photochemical dynamics do not require subcycle precision. Photochemistry will still benefit from the coherent nature of high harmonic generation and the sensitivity of recollision phenomena to electronic and nuclear structural changes.
Recollision is different in a second way. A recolliding electron wavepacket probes its parent and so can probe any correlated motion in the ion. Through correlated measurement, recollision physics shares features with quantum optics. Very fast vibrational motion created by the removal of an electron or very fast electronic motion created by exchange and other interactions between the tunneling electron and the ion become measurable for the first time. 9 , 13
The critical feature of recollision physics is that multiphoton ionization provides access to time and length scales shorter than the laser period and laser wavelength. That feature is a general property of multiphoton interactions—recollision is likely just the first of many new routes to attosecond and subwavelength science.
An obvious frontier is to increase the intensity of the fundamental beam. There is a very long way, at least nine orders of magnitude, before we reach even a practical limit to the light intensity. More-intense light allows higher-energy, broader-bandwidth electrons and photons, and thus enhanced time resolution. I predict that optical pulse durations will fall below 10 attoseconds in the coming years, and spatial resolution well below 1 Å. That would still not even be close to the limit. High-intensity interactions could also lead to a new field of real-time nuclear dynamics in which the recollision bandwidth is sufficiently high to excite a nuclear reaction, and the time-dependent field that probes the dynamics is very strong (see box
Another frontier is solids. In the 1965 paper by Keldysh 1 that launched this field, atoms and condensed media were treated together. But in the decades since, theory and experiments have served mostly to elucidate the behavior of gas-phase atoms. Some of that knowledge can be transferred to large-bandgap solids. That enhanced understanding will guide us to new forms of high-order multiphoton microscopy, high-precision laser surgery, and laser machining.
It will be an exciting future.
Box 1. Light–matter interactions near the ionization threshold
The electric field of a laser pulse exerts a force on all charged particles, be they in atoms, molecules, or solids. The force can approach or exceed the force that binds electrons to atoms or atoms to molecules or solids. Two potentially coexisting cases are interesting:
First, if the light field approaches but doesn’t exceed the binding field, so that the system remains intact, spectral shifts are large enough that the atom’s or the molecule’s spectrum is significantly influenced by the light field. By controlling the light, we gain partial control over the system. Thus it is possible to trap or accelerate an atom or molecule, gate molecular dissociation, and align or spin a molecule, 14 all by controlling the light pulse. (See the article by Ian Walmsley and Herschel Rabitz, PHYSICS TODAY, August 2003, page 43 .)
Second, if the system ionizes, then the ion’s influence over the electron rapidly diminishes. In the gas phase, an IR light pulse can provide subcycle control over the newly freed electron. This is the realm of recollision physics, the general subject of this paper.
The physics discussed here does not necessarily call for large facilities. Many conventional short-pulse tabletop experiments are performed at or near the intensity at which some ionization occurs. But whereas conventional experiments concentrate on the non-ionized part of the sample, tunneling and recollision physics concentrate on the ionizing portion.
Box 2: Collision and optical science
Which of the processes of interest to collision physics can be initiated by laser-induced recollision? If recollision physics could be extended to intensities at which the electron’s energy is relativistic, then the core electron from a highly charged ion could be used to produce a precisely timed recollision energetic enough to initiate a nuclear reaction. That would open a route to time-resolving nuclear decay.
Reaching relativistic energies is not the challenge. Ponderomotive, or oscillating, electron energies up to hundreds of MeV can be reached at the currently accessible intensities of roughly 1022 W/cm2. At such high intensities—and even at intensities several orders of magnitude lower, depending on frequency—the momentum imparted by absorbed photons causes the ionized electron to drift far enough along the propagation direction that on reversing its course it misses the parent ion. Recollision is therefore turned off.
The problem can be overcome using equal-amplitude, counterpropagating, circularly polarized beams of the same handedness. 15 The interfering beams create parallel electric and magnetic fields throughout the focal volume. In such a configuration, the photon momentum in each direction is balanced and—even better—the light’s magnetic field restricts the natural quantum mechanical expansion of the recollision-electron wavepacket in the direction perpendicular to the electric and magnetic fields.
The same light that drives the recollision and thereby initiates a nuclear reaction can drive what’s known as an attosecond streak camera 3 to measure when decay fragments appear in the continuum. Although many experimental hurdles remain, the path extending ultrafast measurement into the realm of nuclear physics appears ready to be paved.
I thank the National Research Council Canada and the University of Ottawa for having created a dynamic environment in which recollision physics could germinate. I gratefully acknowledge funding from the NRCC, the Natural Sciences and Engineering Research Council of Canada, the Canadian Institute for Photonic Innovations, the Canada Foundation for Innovation, the Ontario Ministry of Research and Innovation, the US Air Force Office of Scientific Research, and the US Army Research Office.
References
1. L. V. Keldysh, Sov. Phys. JETP https://doi.org/SPHJAR 20, 1307 (1965).
2. P. B. Corkum, Phys. Rev. Lett. https://doi.org/PRLTAO 71, 1994 (1993);
N. H. Burnett, P. B. Corkum, J. Opt. Soc. Am. B https://doi.org/JOBPDE 6, 1195 (1989).3. P. Agostini, L. DiMauro, Rep. Prog. Phys. https://doi.org/RPPHAG 67, 813 (2004);
P. B. Corkum, F. Krausz, Nat. Phys. 3, 381 (2007);
F. Krausz, M. Ivanov, Rev. Mod. Phys. https://doi.org/RMPHAT 81, 163 (2009).4. X. M. Tong, Z. X. Zhao, C. D. Lin, Phys. Rev. A https://doi.org/PLRAAN 66, 033402 (2002).
5. M. Ivanov, M. Spanner, O. Smirnova, J. Mod. Opt. https://doi.org/JMOPEW 52, 165 (2005).
6. T. Zuo, A. D. Bandrauk, P. B. Corkum, Chem. Phys. Lett. https://doi.org/CHPLBC 259, 313 (1996);
M. Lein, J. Marangos, P. Knight, Phys. Rev. A https://doi.org/PLRAAN 66, 051404–R (2002).7. M. Meckel et al., Science https://doi.org/SCIEAS 320, 1478 (2008);
M. Gertsvolf et al., Phys. Rev. Lett. https://doi.org/PRLTAO 101, 243001 (2008).8. M. Lewenstein et al., Phys. Rev. A https://doi.org/PLRAAN 49, 2117 (1994).
9. O. Smirnova et al., Nature https://doi.org/NATUAS 460, 972 (2009).
10. J. Itatani et al., Nature https://doi.org/NATUAS 432, 867 (2004);
S. Haessler et al., Nat. Phys. 6, 200 (2010).11. D. Shafir et al., Nat. Phys. 5, 412 (2009);
H. Niikura et al., Phys. Rev. Lett. https://doi.org/PRLTAO 105, 053003 (2010).12. Y. Mairesse et al., Phys Rev. Lett. https://doi.org/PRLTAO 100, 143903 (2008);
H. Wörner et al., Nature https://doi.org/NATUAS 466, 604 (2010).13. H. Niikura et al., Nature https://doi.org/NATUAS 421, 826 (2003);
S. Baker et al., Science https://doi.org/SCIEAS 312, 424 (2006).14. H. Sakai et al., Phys. Rev. A https://doi.org/PLRAAN 57, 2794 (1998);
H. Stapelfeldt, T. Seideman, Rev. Mod. Phys. https://doi.org/RMPHAT 75, 543 (2003).15. N. Milosevic, P. B. Corkum, T. Brabec, Phys. Rev. Lett. https://doi.org/PRLTAO 92, (2004).
More about the authors
Paul Corkum is a professor of physics at the University of Ottawa and director of attosecond science at the National Research Council Canada, both in Ontario.





{kind=link}
{kind=link}
{kind=link}
{kind=link}