Macromolecular phasing
DOI: 10.1063/1.2195315
Modern molecular biophysics is built on the twin pillars of genetic engineering and macromolecular structure determination. And x-ray crystallography is foremost among the methods used to determine the atomic-scale structure of macromolecules—very large molecules with sizes typically ranging from 50 to 1000 Å. X-ray crystallography played a pivotal role in the science of the 20th century and has led directly to no fewer than eight Nobel prizes.
In 1912 Max von Laue explained how the periodic lattice of a crystal scatters an incident beam of x rays in specific directions in space, and, with Walter Friedrich and Paul Knipping, he discovered the first x-ray diffraction pattern. Practical x-ray diffraction crystallography dates back to the father-and-son team of William and Lawrence Bragg, who determined the crystal structure of sodium chloride (NaCl) in 1914, little more than a year after von Laue and his colleagues reported their discovery. By analyzing the directions and the intensities of the Bragg reflections—that is, the scattered beams—one can, in principle, figure out the spatial arrangement of the atoms inside the crystal.
In practice, the inversion from measured diffraction intensities to atomic structure is not as straightforward as one might think: The intensity relates to the amplitude of a scattered wave, but not to its phase relative to the other scattered waves. Without those phases, one simply does not know how to add all scattered waves together to retrieve the original structure, especially when that structure is a macromolecule such as a protein or virus. That difficulty is called the phase problem of x-ray crystallography, and its solution is often referred to as phasing a structure (see reference and the article by Keith Nugent, David Paganin, and Tim Gureyev, Physics Today, August 2001, page 27 ).
The crystal structure may be expressed in terms of the electron density ρ(r) as a function of position r in the unit cell of the crystal; from that density one can infer the positions of the atoms. The electron density is related to the Bragg reflection amplitudes, or structure factors, F H, which are Fourier transforms of the density function:
where H indexes the reflections manifested in the diffraction pattern.
A structure factor F H = |F H |exp(iφ H ) is a complex number with magnitude |F H | and phase φ H. Detectors measure intensities proportional to |F H |2; phase information is lost. The
Over the past century, crystallographers have developed powerful techniques for recovering phase information. Those phasing innovations, along with advances in x-ray sources and detectors, computing power, and techniques for handling crystals, have revolutionized how structures are determined by x-ray crystallography. Figure 1 illustrates the steady progress in solving the structures of ever-larger molecules. New techniques are constantly being developed, including some that will allow scientists to map out such nonperiodic materials as cells.

Figure 1. Ever-larger crystalline structures have been solved since the sodium chloride (NaCl) structure was determined in 1914. Phasing methods, designed to recover the phases of scattered x rays, have been key to the success of crystal-based structural science. The straight line through the data is to guide the eye. An asymmetric unit is a subset of a crystal’s unit cell, which comprises asymmetric units related by symmetry operations. The submicrometer scale of the most complex objects in the figure is near the border between what one can see directly with microscopic and imaging techniques and what one can study only with diffraction and scattering.
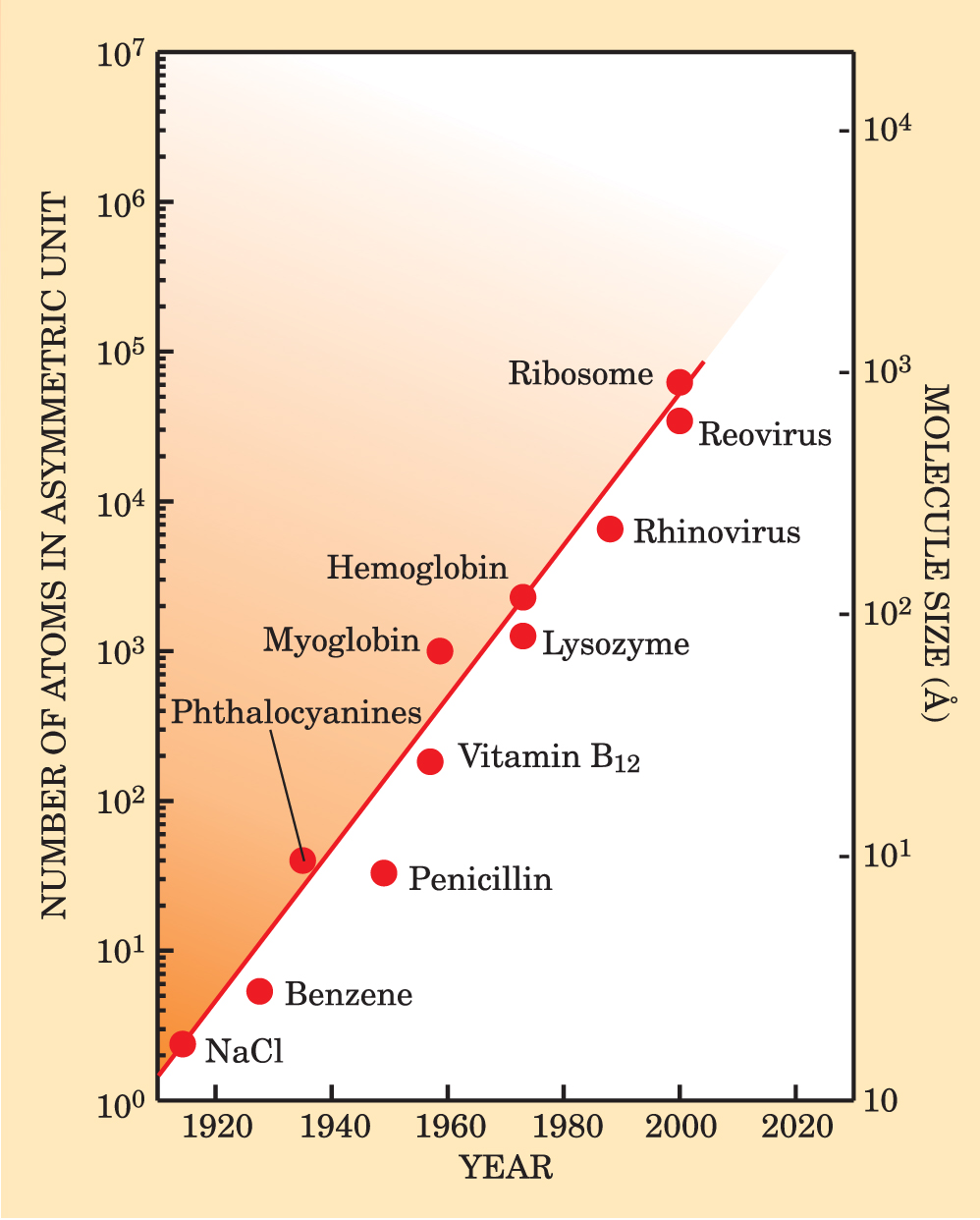
Some 50 years ago, when John Kendrew in Max Perutz’s group at Cambridge University solved the first protein structure, myoglobin (a protein of about 2600 atoms found in muscles that store oxygen), his success was the culmination of more than 10 years of patience and perseverance during which he went through 110 crystals to measure more than 250 000 Bragg reflections. Today, comparably sized protein structures are determined in just a few hours at modern synchrotron facilities with the help of powerful phasing programs.
Established methods
Early on, crystallographers solved the structures of small molecules by exploiting the fact that Fourier transforming the measured intensity gives the charge density autocorrelation function, also known as the Patterson function. In practice, the Patterson function gives a map of the scattering crystal’s interatomic vectors. After considering all possible vector arrangements in trial structures, one can find the structure that best fits the experimentally determined Patterson function. As the structures get larger and more complex, however, that simple trial-and-error method becomes impractical due to the rapid growth in the number of interatomic vectors.
Over the years, researchers have developed more powerful phasing methods, which can be grouped generally into three categories. The first includes mathematical techniques, the second includes phasing techniques based on binding an atom or molecular group to the crystal structure, and the third encompasses those techniques that exploit known structures.
Among the mathematical techniques available to crystallographers are ab initio direct methods, 2 including those that won Herbert Hauptman and Jerome Karle the 1985 Nobel Prize in Chemistry. Direct methods combine an overdetermined number of intensity measurements with a probability distribution of possible phases and with such physical constraints as the highly peaked electron densities at atomic locations. Direct methods are very powerful for small molecules, and at present the crystallographic community is tremendously interested in extending those methods to macromolecular crystal structures. The surge of recent interest has resulted primarily from three factors: the ability to obtain atomic-resolution (below 1.2 Å) data in favorable cases at modern synchrotron sources, the development of Shake & Bake 3 and other advanced computer codes, and the ability to combine direct methods with the techniques of isomorphous replacement and anomalous scattering.
The phase problem
Crystallographic measurements give the amplitudes of structure functions F, which represent Fourier components of the electron density. But they do not give phases φ, and without those phases one cannot unambiguously determine the density. To see how that difficulty plays out in detail, suppose that three Bragg reflections, (100), (200), and (300), have measured amplitudes |F(100)| = |F(200)| = |F(300)| = 0.5, representing the Fourier components of the electron density in the x direction. Even if the phases of the structure functions are limited to 0 or π, as is the case when the crystal repeat unit has an inversion symmetry, and even if two of the phases are known, the ambiguity in the third phase can have a marked effect on the deduced electron density. In the illustrations at right, the zeroth order (000) reflection corresponds to a constant added to the density, and the phases for the (200) and (300) reflections have been set equal to 0. A phase of 0 for F(100) gives rise to the blue electron density whereas a phase of π yields the distinctly different green density.

Heavy subunits
Among the early techniques based on binding were single and multiple isomorphous replacement (SIR and MIR). In those techniques, heavy atoms, that is, atoms with a high atomic number, are bound to specific sites on the protein. If the addition does not alter the structure of the protein, then the diffractions of the atom and of the protein interfere in a way that can be used to extract the phases. Because the diffraction intensity of an atom scales quadratically with its atomic number, the contribution of a single heavy atom such as gold can be readily observed, even when the heavy atom is attached to a protein consisting of thousands of atoms with low atomic numbers. Isomorphous replacement techniques are still widely used, although adding the heavy atom may be difficult without altering the protein structure.
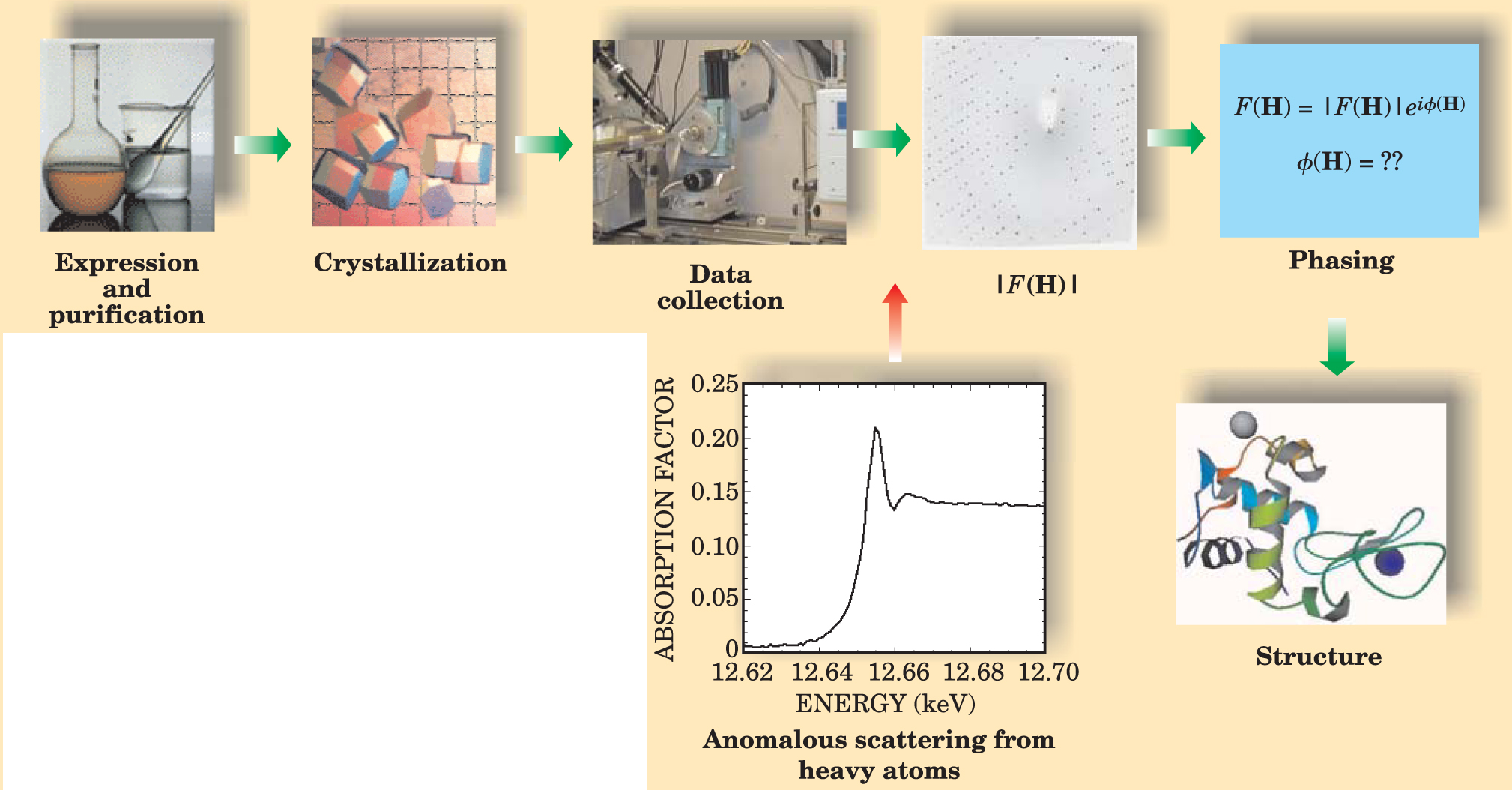
Before intense, wavelength-tunable synchrotron x-ray sources became available, phasing methods had to use the characteristic line radiation from x-ray tubes, since monochromatized continuum bremsstrahlung radiation was usually too weak for practical macromolecular diffraction work. Synchrotron radiation has catalyzed the development of several alternative methods of solving the phase problem. For more than a decade now, the multiple anomalous diffraction (MAD) method has been commonly used in protein crystallography (see reference and the article by Wayne Hendrickson, Physics Today, November 1995, page 42 ). The word “anomalous” refers to the shift in a scattered wave, a shift that results when the incident x-ray energy is close to an atomic absorption edge. The MAD technique uses the amplitudes and anomalous phase shifts from diffraction at two or three wavelengths near an absorption edge of specific elements intrinsic to or grown into the protein. The most commonly used anomalous scatterer is selenium, which can be substituted for sulfur in methionine, one of 20 amino acid residues that make up proteins. Figure 2 displays a typical anomalous-scattering spectrum along with several of the steps used in determining a protein structure.

Figure 2. Protein crystallography typically requires several steps from sample preparation to structure determination. The most common phasing technique is based on anomalous scattering near an absorption edge of heavy atoms incorporated in a structure. The spectrum here displays a typical anomalous-scattering energy scan near the selenium K edge.
The explosive growth in structural biology itself has been the result of many important innovations in MIR, SIR, and MAD phasing of large macromolecular entities. Those innovations include the use of cluster compounds and the development of alternative anomalous scatterers. Improvements in modern direct methods and other algorithms have allowed crystallographers to find large numbers of anomalous atom sites in large selenoprotein structures, to give just one example. Anomalous-scattering methods are now the workhorses for solving novel structures in macromolecular crystallography. Some proteins, however, resist the insertion of an anomalous atom. Complex mammalian proteins, in particular, are frequently resistant.
Molecular replacement
A third phasing technique has been widely used for target macromolecules that contain a known subunit or that can be related to a known structure. 5 To use the method, called molecular replacement, one needs an observed diffraction pattern for the target and the atomic coordinates of the probe—that is, of the subunit or related structure. After determining the position of the probe within the unit cell of the target crystal, one can calculate approximate target phases.
Six coordinates—three rotational and three translational—are needed if one is to correctly establish the position of the probe. In principle, a search on the six variables will reveal the position that gives the best agreement between observed and calculated structure factors. However, such a search would be computationally too demanding. The key to efficiently locating the probe is to separate the rotational and translational variables. The Patterson function maps interatomic vectors and is independent of any choice of origin, so it is well suited to the task. It allows one to reduce the six-dimensional search to two 3D searches. The first determines the correct orientation of the probe, and the second situates the correctly oriented molecule within the unit cell.
Sometimes a crystallographic asymmetric unit contains two or more identical subunits. In that case, the molecular replacement method can be used to find the operation—a non-crystallographic symmetry (NCS)—that superimposes one subunit on the other. By exploiting the redundancy of NCS and a technique called molecular averaging, one can obtain dramatically improved phases and resolution. Many virus structures have yielded to the molecular averaging method.
New techniques
Established phasing methods face two problems: the sensitivity of samples to radiation damage and the difficulty of incorporating foreign atoms into samples. Many newly emerging phasing techniques address those difficulties. Nowadays crystallographers can select from a menu of techniques, sometimes in combination.
The MAD technique, described earlier, involves measuring the diffraction intensities at multiple x-ray wavelengths near an absorption edge of an incorporated atom such as Se. Since most protein crystals are easily damaged by radiation, it is desirable to reduce the number of data sets required to solve a structure. The single anomalous diffraction (SAD) method 6 has enabled such a reduction. With it, protein structures have been solved using a diffraction data set from a single x-ray wavelength near the absorption edge of the anomalous scatterer.
Even assuming that the measured amplitudes and the calculated amplitude and phase contributions of the anomalous scatterer are error free, there remains, in SAD experiments, an inherent twofold ambiguity in the estimation of the protein phase for each reflection. 1,6 One way to resolve the ambiguity is to choose the phase closer to that of the anomalous scatterer. Wayne Hendrickson and Martha Teeter used that method to determine the structure of crambin, a small hydrophobic protein. Bi-Cheng Wang developed a different method to resolve the phase ambiguity; his iterative density-modification technique uses protein–solvent boundary information. Similar density-modification schemes have been implemented in several phasing software packages.
Direct methods, too, have been used for many years to break the SAD phase ambiguity. 2 For example, Ian Harvey and colleagues, following earlier work by Hai-Fu Fan and Yuan-Xin Gu, solved the structure of rusticyanin, an acid-stable copper protein. 7 Their procedure combined SAD data from a native crystal with direct methods and density modification. Recent applications include SAD phasing with Se atoms grown into a protein, or with xenon, heavy halide atoms, or other small molecule complexes diffused into the crystal.
Envelope phasing is a promising technique that does not require the presence of incorporated atoms; it instead utilizes the low-resolution shape, or envelope, of the protein. The envelope may be determined by small-angle x-ray scattering of a protein in solution, electron-microscope images of isolated molecules, or the anomalous-solvent-contrast method developed by Roger Fourme and colleagues. 8 To utilize the envelope information, a crystallographer must determine the known envelope’s orientation and position in a crystallographic unit cell: In that regard, the envelope phasing technique is similar to molecular replacement, for which the Patterson function is conventionally used to determine the probe’s position. However, Patterson searches are usually not appropriate in the context of envelope phasing because the density inside the envelope is uniform—the intra-envelope Patterson vectors cannot be discriminated.
One of us (Hao) has developed a method to perform a simultaneous 6D search on orientation and translation to find the best match between structure factors determined by experiment and those calculated from a presumed location of the envelope in the unit cell. 9 In the case of a macromolecule lacking NCS, one must conduct a full 6D search. For a molecule with NCS, one can perform the search in two separate stages. First, a rotation search yields two Eulerian angles for the NCS axis of the molecular envelope. Knowledge of the NCS-axis orientation reduces the 6D search to a 4D search for a third Eulerian angle and three translation parameters. And that reduction significantly cuts the calculation time needed to locate the envelope.
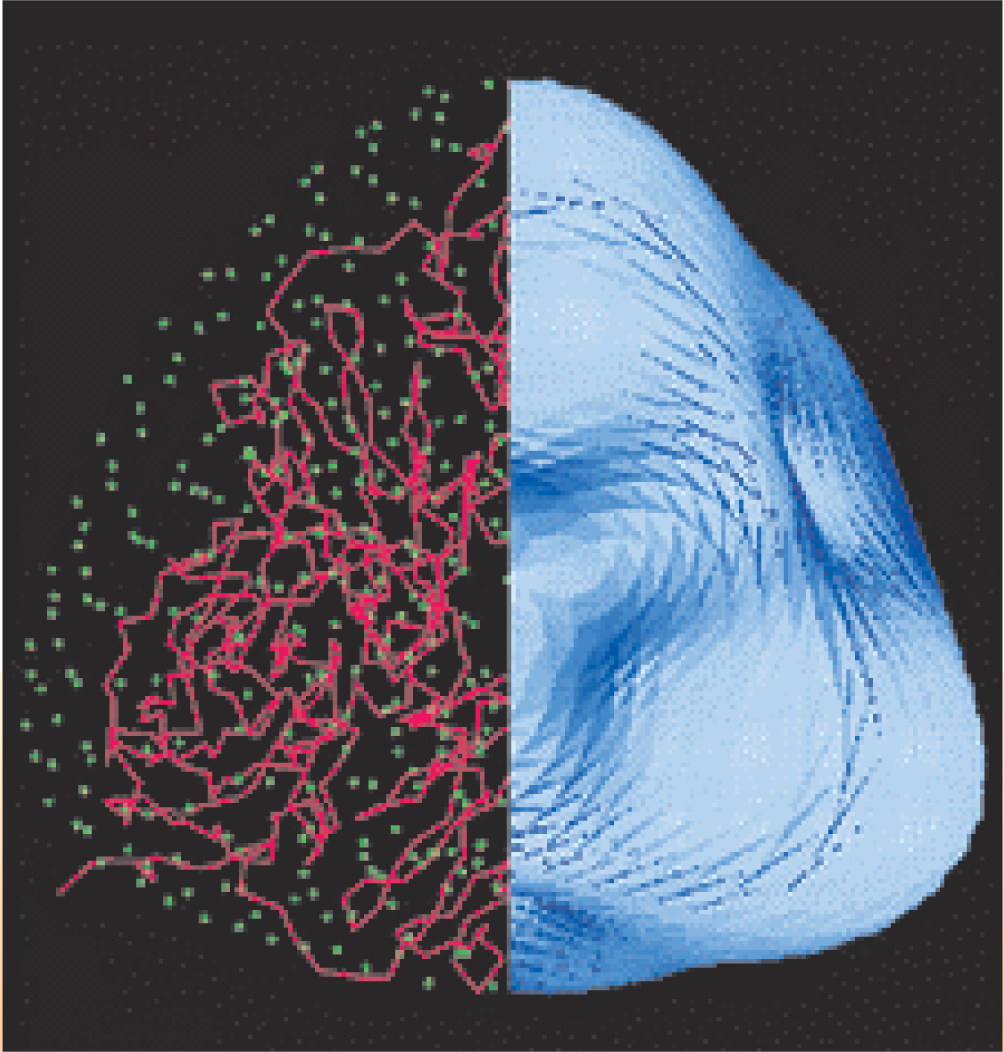
As illustrated in figure 3, the low-resolution phases calculated from the correctly positioned molecular envelope can be a good starting point for a variety of phase-extension techniques that ascertain a macromolecule’s internal structure with greater resolution; such techniques include maximum-entropy and density-modification methods.

Figure 3. Determining the envelope of a macromolecule can serve as a first step toward detailed structure determination. In this illustration, the right-hand side shows the envelope of the enzyme nitrite reductase. Combining knowledge of the envelope’s position with other phasing techniques yields the detailed structure shown on the left.
(Adapted from ref. 9, Q. Hao.)
Three-beam phasing
More than half a century ago, William Lipscomb proposed a way to obtain the phases of the Fourier components directly from diffraction experiments. 10 His idea of three-beam Bragg diffraction is based on the interference between simultaneously excited Bragg waves from two different sets of atomic planes in the crystal. The concept is analogous to holography, which uses a reference wave.
A convenient and efficient way to collect a large number of three-beam diffraction profiles in protein crystals is to use a reference-beam diffraction geometry (RBD). Figure 4 shows an application of the RBD that combines the principle of three-beam diffraction with the most common crystallographic data collection technique, the oscillating crystal method, in which the crystal is rotated about an axis. 11 Usually, the axis is perpendicular to the incoming x-ray beam, but in the RBD, the axis is tilted by the Bragg angle θ G of a strong reference reflection G. As a result, the reflection G remains fully excited as the crystal is rotated. In principle, the intensities of all Bragg reflections recorded on an area detector as the crystal is rotated can be influenced by the interference with the reference reflection, so one can collect many three-beam profiles simultaneously. In practice, in addition to rotating the crystal, one varies the tilt angle θ to obtain complete interference profiles. From those profiles, a crystallographer can extract relative structure-factor phases, called triplet phases, using recently developed phase-sensitive x-ray diffraction theories.

Figure 4. In the reference-beam diffraction geometry (RBD), a crystal sample is rotated about an axis corresponding to a strong reference reflection G. (a) The interference between the red reflections from the reference plane and blue reflections from another plane is the physical phenomenon that allows for the extraction of phase information. The orientation of the crystal is described by a tilt angle θ with respect to a reference axis (dotted line). A vanishing tilt angle indicates that the crystal is oriented perpendicular to the incoming beam (purple); the angle θ G corresponds to the RBD. (b) X-ray area detectors collect phase-sensitive data at many different θ. The plot below the five sample spectra illustrates the intensity of the reference beam at different tilt angles; the red curve is a Gaussian fit. The graphs to the right show the intensity of three diffraction peaks as a function of tilt angle. From such profiles one can obtain relative structure-factor phases.

In the past few years, several works have focused on phasing algorithms that make use of measured triplet phases. 12 For example, Charles Weeks and coworkers have incorporated triplet phases into existing direct methods, and Qun Shen and Jun Wang have developed a recursive algorithm to retrieve the structure-factor phases. Alexei Soares and colleagues have combined sophisticated modeling techniques with experimentally measured three-beam phases to refine the structure of rhombohedral insulin, a protein.
Before the three-beam phasing technique can be widely adopted, experimenters will need to overcome several challenging obstacles; those include obtaining crystals with very high perfection and precisely aligning them. If those issues are resolved, the three-beam method may provide the necessary phase information for protein crystallography without the need to incorporate extra atoms into a native protein structure.
Nonperiodic specimens
The success of structural science is based mainly on the use of crystalline specimens. However, not all materials can be crystallized; membrane proteins and larger multiprotein macromolecular assemblies are particularly difficult.
In the early 1980s, David Sayre proposed using coherent x-ray diffraction as an imaging technique to determine the electron density in a noncrystalline single-particle specimen. 13 The idea is to measure the far-field coherent x-ray diffraction pattern, which is the absolute square of the Fourier transform F(Q) of the density, and to reconstruct the phases of the measured diffraction amplitudes. Scientists have made considerable progress in the past few years, and several x-ray experiments have demonstrated the feasibility of the technique, which can potentially allow x-ray imaging to angstrom dimensions. Even so, coherent-beam methods are in a relatively early stage of development and scientists will need to overcome many challenges if those methods are to become practical.
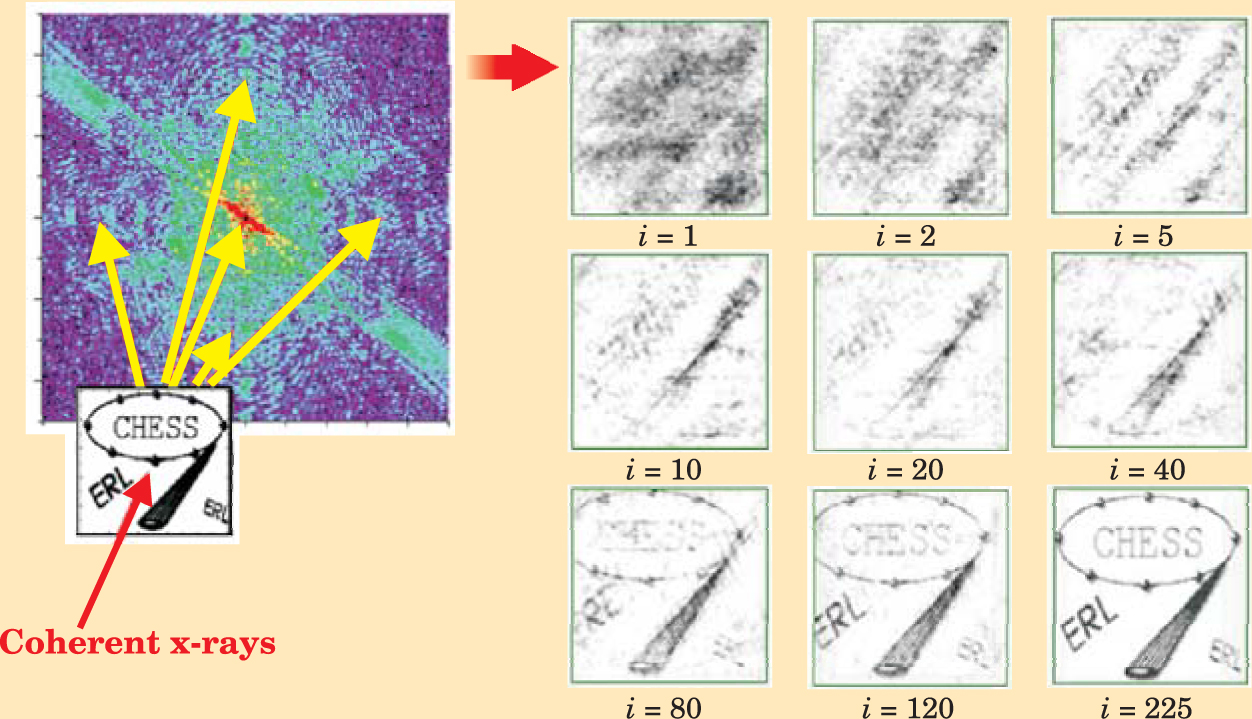
Coherent diffraction imaging of noncrystalline specimens is analogous to x-ray crystallography, but with two important differences. First, the Fourier transform F(Q), which plays the role of the structure function, is a continuous function; crystals yield discrete Bragg peaks. Thus iterative phasing algorithms for phase retrieval and structure determination may be used (see figure 5), 14 provided that one oversamples, that is, measures the continuous diffraction pattern at angular intervals finer than the spacing that would separate the Bragg peaks if the sample were crystallized. Second, coherent diffraction imaging requires an intense coherent x-ray or electron beam to overcome the lack of periodicity in the specimen and preserve the phase information in the diffraction pattern. At today’s synchrotron x-ray sources, the transverse coherent portion of a typical undulator hard-x-ray beam is less than 1% so coherent diffraction experiments are difficult. That is one of the main reasons that the crystallographic community is actively discussing several proposals for a fully or mostly coherent x-ray source, such as an x-ray free-electron laser (XFEL) or an energy-recovery linear accelerator (ERL). 15

Figure 5. Coherent diffraction imaging, in combination with iterative phasing methods, can solve the structures of nonperiodic samples. In this simulation, the goal is to reconstruct the CHESS image on the lower left, made from 2894 gold atoms in a 10 nm × 10 nm square. The diffraction intensities are indicated in color; purple indicates low intensity, red indicates high. To the right are examples of reconstructed images obtained with iterative phasing methods. As the number of iterations i increases, the image-reconstruction quality improves.
(Adapted from ref. 16, Q. Shen et al.)
The intense x-ray or electron beams required for coherent imaging cause radiation damage, which ultimately sets the achievable spatial resolution. For single-particle biological specimens, radiation damage may limit the resolution to a few nanometers, 16 but more resistant specimens, including hard materials, may allow for nearly atomic resolution. Further study is required for both biological and material cases, especially for frozen or freeze-dried biological specimens.
Several techniques are being proposed to overcome the radiation-damage problem for biological specimens and allow crystallographers to get close to atomic resolution. One possibility is to use a single very short pulse from an XFEL to record a diffraction pattern before prompt photoelectron emission causes the molecule to explode due to Coulomb repulsion. However, at least for the XFELs currently being projected, the intensity from a single XFEL pulse would not be strong enough to record a high-resolution diffraction pattern. Many Poisson-noise-limited patterns from repeated pulses would need to be sorted and combined to provide statistically significant signals at atomic resolution. Theoretical models suggest that such experiments present a considerable challenge to XFEL designers: The laser pulses need to be shorter than 2–4 fs.
Another idea is to perform a coherent diffraction experiment on a continuous stream of frozen macromolecules. 17 In such experiments, intense optical laser fields align the molecules, although the alignment may need to be aided by optically anisotropic entities bound to the molecule. The advantage of the continuous-stream approach is that many molecules are in the beam at a time, which improves the statistics of the recorded images of the scatter. Continuous-stream experiments may be best suited to a continuous coherent x-ray or electron beam source such as an ERL.
Coherent diffraction holds out the prospect of atomic-resolution “crystallography without crystals,” but it has perhaps even greater potential as a tool for studying frozen or freeze-dried cellular structures or other larger biological assemblies (see figure 6). Although the technique would not achieve atomic spatial resolution for those larger, frozen structures, it would yield resolutions far better than those achievable today with confocal lasers (about 100 nm) and with soft-x-ray microscopes (about 20 nm). The many advances in coherent diffraction are illustrated by successes in phasing Escherichia coli bacteria, nanocrystallites, and, as shown in figure 7, frozen yeast cells. 18 Today, structural science is based mostly on crystals. But diffractive-imaging experiments with coherent x-ray and electron sources, in conjunction with lens-based cryogenic-x-ray and electron microscopy, promise to open up the field to include studies of nonperiodic objects in cell biology and materials sciences, where exact copies of the specimen may not exist.

Figure 7. The phasing of a freeze-dried yeast cell illustrates the power of coherent diffraction. (a) This diffraction pattern (with a false color scale) was obtained with coherent, soft 750-eV x rays at Lawrence Berkeley National Laboratory’s Advanced Light Source. (b) In this image of the yeast cell, brightness gives magnitude, and hue illustrates phase. The spatial resolution is better than 30 nm. Phase retrieval was accomplished with an iterative method.
(Images courtesy of Veit Elser and Pierre Thibault at Cornell University and their collaborators at Stony Brook University and LBNL; see ref. 18, D. Shapiro et al.)


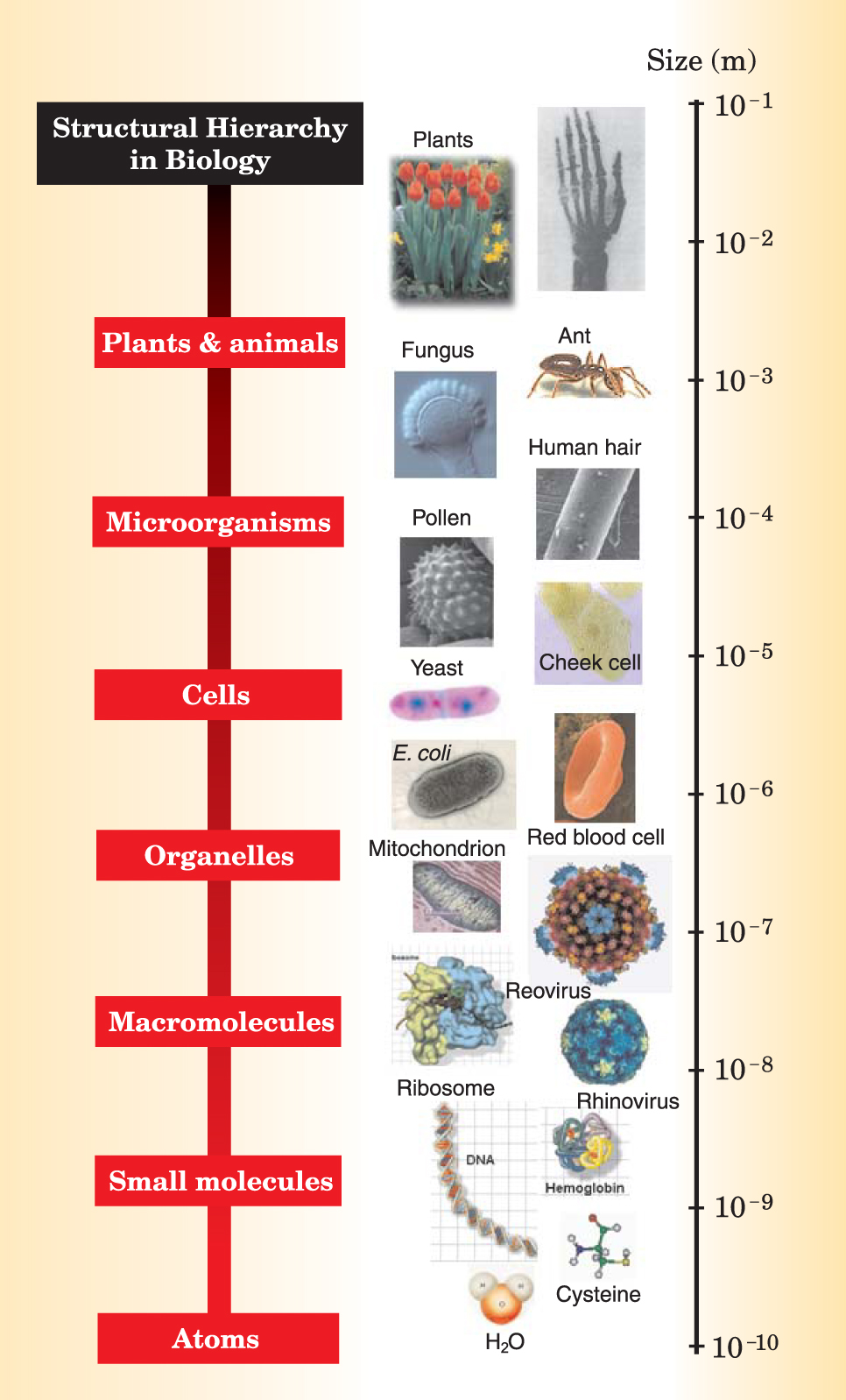
Figure 6. Biological systems have a hierarchy of structures that range over many orders of magnitude in size.
Since the birth of x-ray crystallography nearly a century ago, the technique has helped scientists determine ever more complicated structures. An essential component in solving all but the simplest of them is phasing. In the future, phasing complex structures, in both crystalline and noncrystalline forms, will continue to play an important role in biological and materials-science investigations at both fundamental and functioning levels.
References
1. T. L. Blundell, L. N. Johnson, Protein Crystallography, Academic Press, New York (1976).
2. See, for example, the review by H. Hauptman, Curr. Opin. Struct. Biol. 7, 672 (1997) https://doi.org/10.1016/S0959-440X(97)80077-2 .
3. R. Miller et al., Science 259, 1430 (1993) https://doi.org/10.1126/science.8451639 .
4. W. A. Hendrickson, Science 254, 51 (1991) https://doi.org/10.1126/science.1925561 .
5. M. G. Rossmann, The Molecular Replacement Method, Gordon & Breach, New York (1972).
6. W. A. Hendrickson, M. M. Teeter, Nature 290, 107 (1981); https://doi.org/10.1038/290107a0
B. C. Wang, Methods Enzymol. 115, 90 (1985); https://doi.org/10.1016/0076-6879(85)15009-3
Z. Dauter, M. Dauter, E. Dodson, Acta Cryst. D 58, 494 (2002) https://doi.org/10.1107/S090744490200118X .7. H.-F. Fan, Y.-X. Gu, Acta Cryst. A 41, 280 (1985); https://doi.org/10.1107/S0108767385000599
I. Harvey et al., Acta Cryst. D 54, 629 (1998) https://doi.org/10.1107/S0907444998005423 .8. W. Shepard, R. Kahn, M. Ramin, R. Fourme, Acta Cryst. D 56, 1288 (2000) https://doi.org/10.1107/S0907444900009574 .
9. Q. Hao et al., Acta Cryst. D 55, 243 (1999); https://doi.org/10.1107/S0907444998011342
Q. Hao, Acta Cryst. D 57, 1410 (2001) https://doi.org/10.1107/S0907444901009374 .10. W. N. Lipscomb, Acta Cryst. 2, 193 (1949) https://doi.org/10.1107/S0365110X49000515 .
For reviews see, for example, R. Colella, Comments Cond. Mat. Phys. 17, 199 (1995);
S. L. Chang, X-Ray Multiple-Wave Diffraction, Springer-Verlag, New York (2004).11. Q. Shen, Phys. Rev. Lett. 80, 3268 (1998) https://doi.org/10.1103/PhysRevLett.80.3268 .
For a review see Q. Shen, Adv. Imag. Elec. Phys. 135, 69 (2005) https://doi.org/10.1016/S1076-5670(05)34002-X .12. C. M. Weeks et al., Acta Cryst. A 56, 280 (2000); https://doi.org/10.1107/S0108767300000829
Q. Shen, J. Wang, Acta Cryst. D 59, 809 (2003); https://doi.org/10.1107/S0907444903003664
A. Soares et al., Acta Cryst. D 59, 1716 (2003) https://doi.org/10.1107/S0907444903015403 .13. D. Sayre, in Imaging Processes and Coherence in Physics, M. Schlenker et al., eds., Springer-Verlag, New York (1980), p. 229 https://doi.org/10.1007/3-540-09727-9_82 .
For a recent review see J. Miao et al., Ann. Rev. Biophys. Biomol. Struct. 33, 157 (2004) https://doi.org/10.1146/annurev.biophys.33.110502.140405 .
For related work using electrons, see J. M. Zuo et al., Science 300, 1419 (2003) https://doi.org/10.1126/science.1083887 .14. R. W. Gershberg, W. O. Saxton, Optik 25, 237 (1972);
J. R. Fienup, Appl. Opt. 21, 2758 (1982); https://doi.org/10.1364/AO.21.002758
J. Miao et al., Nature 400, 342 (1999); https://doi.org/10.1038/22498
S. Marchesini et al., Phys. Rev. B 68, 140101 (2003); https://doi.org/10.1103/PhysRevB.68.140101
V. Elser, J. Opt. Soc. Am. A 20, 40 (2003) https://doi.org/10.1364/JOSAA.20.000040 .15. See, for example, T. Tschentscher, Proc. SPIE 4500, 1 (2001); https://doi.org/10.1117/12.452963
J. Arthur et al., Linac Coherent Light Source (LCLS) Conceptual Design Report, Stanford University, Stanford, CA (April 2002), available at http://www.slac.stanford.edu/pubs/slacreports/slac-r-593.html ;
S. M. Gruner et al., Rev. Sci. Instrum. 73, 1402 (2002) https://doi.org/10.1063/1.1420754 .16. S. Marchesini et al., Opt. Express 11, 2344 (2003); https://doi.org/10.1364/OE.11.002344
Q. Shen, I. Bazarov, P. Thibault, J. Synch. Rad. 11, 432 (2004) https://doi.org/10.1107/S0909049504016772 .17. J. C.H. Spence, R. B. Doak, Phys. Rev. Lett. 92, 198102 (2004) https://doi.org/10.1103/PhysRevLett.92.198102 .
18. J. Miao et al., Proc. Natl. Acad. Sci. USA 100, 110 (2003); https://doi.org/10.1073/pnas.232691299
G. J. Williams et al., Phys. Rev. Lett. 90, 175501 (2003); https://doi.org/10.1103/PhysRevLett.90.175501
D. Shapiro et al., Proc. Natl. Acad. Sci. USA 102, 15343 (2005) https://doi.org/10.1073/pnas.0503305102 .
More about the authors
Qun Shen, a staff scientist at the Cornell High Energy Synchrotron Source (CHESS) at Cornell University in Ithaca, New York, is currently a senior physicist at the Advanced Photon Source at Argonne National Laboratory in Argonne, Illinois, and an adjunct professor of materials science at Northwestern University in Evanston, Illinois. Quan Hao is the director of MacCHESS, the macromolecular diffraction facility at CHESS. Sol Gruner is the director of CHESS and a member of Cornell’s physics department.
Qun Shen, 1 Cornell High Energy Synchrotron Source (CHESS), Cornell University, Ithaca, New York, US .
Quan Hao, 1 Cornell High Energy Synchrotron Source (CHESS), Cornell University, Ithaca, New York, US .
Sol M. Gruner, 1 Cornell High Energy Synchrotron Source (CHESS), Cornell University, Ithaca, New York, US .





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}