Diatomic molecules, a window onto fundamental physics
DOI: 10.1063/PT.3.3020

For a long time most atomic physicists were trained in accordance with 1981 Nobel laureate Arthur Schawlow’s dictum, “A diatomic molecule is a molecule with one atom too many.” Compared with atoms, even the simplest molecules seem dauntingly complicated. Their internal vibrational and rotational (collectively called “rovibrational”) motions and their lack of spherical symmetry introduce structural features completely absent in atoms. That complexity makes the quantum mechanical properties of molecules far more difficult to manipulate, control, and measure than in their simpler atomic counterparts. Nevertheless, during the past several years, physicists have begun to work with molecules in high-precision experiments designed to probe some of the most fundamental features of physical law.
Such precision measurements have long played an important role in atomic physics. For example, experiments have looked for violations of spatial-inversion symmetry, or parity (P), to investigate the possible existence of new particles analogous to the Z gauge boson. Other experiments have searched for a permanent electric dipole moment (EDM) in a particle, which would provide evidence of CP-violating forces needed to explain the dominance of matter over antimatter in the universe (CP denotes the joint operation of charge conjugation and spatial inversion). And comparisons of atomic clocks based on different types of atomic energy levels have explored whether the fundamental constants of nature actually vary in time or space. (For Physics Today coverage, see April 1997, page 17 ; the article by Norval Fortson, Patrick Sandars, and Steve Barr, June 2003, page 33 ; and the article by Maxim Pospelov and Michael Romalis, July 2004, page 40 .)
Remarkably, the additional degrees of freedom in molecules can greatly enhance the measurable signals that manifest the underlying fundamental physics—often to many orders of magnitude above the analogous signals in atoms. 1 Furthermore, recent years have seen significant advancements in experimental methods for the control and measurement of molecular energy levels (see reference and the article by Debbie Jin and Jun Ye, Physics Today, May 2011, page 27 ). Those techniques were developed largely to enable the use of ultracold molecules to address questions in quantum many-body physics, quantum chemistry, and quantum information processing. Those same methods, however, are also making it possible to exploit the inherent advantages of molecules for testing deep questions related to particle physics, nuclear physics, and cosmology.
Level with me
The structural complexity of molecules gives them a denser spectrum of energy levels than atoms have. For simple molecules, as the
In more complex molecules, additional energy scales can arise. An example is the hydrogen peroxide (HOOH) molecule illustrated in figure 1. In this case, the new energy scale arises from the torsional motion of one H atom relative to the plane defined by the OOH structure. That motion changes the distance between nearby H atoms, and so its characteristic energy is the vibrational energy. Classically, that energy would be minimized in the two mirror-image configurations for which the rotation angle θ = ±θ0, with θ0 roughly equal to π/2. Quantum mechanically, however, the hydrogen atom can tunnel through the barrier at θ = 0; energy eigenstates are symmetric and antisymmetric combinations of the two classical minimum-energy configurations. The small energy splitting between the two eigenstates is proportional to the tunneling rate, which is exponentially suppressed relative to the natural vibrational frequency. For the particular case of HOOH, the torsional-doublet splitting is comparable to the molecule’s rotational energy.

Figure 1. Mirror images and tiny energy splittings. Torsional motion in hydrogen peroxide (HOOH; left panel) leads to two mirror-image configurations, with θ roughly equal to ±π/2, that have the minimum classical energy. Because of tunneling through the barrier that separates those configurations, would-be degenerate vibrational levels are split, as indicated by the red and green lines in the spectrum shown to the right. In general, the splitting is much smaller than the vibrational-energy scale; in HOOH it is comparable to the energy required to impart a single ℏ unit of rotational angular momentum to the molecule.

Even diatomic molecules can display a similar effect, if they have an electron orbiting the internuclear axis with a nonvanishing angular-momentum component Ω along the axis. In that case, a mirror-image state with the electron orbiting in the opposite direction would nominally have the identical energy. However, since the entire molecule is free to rotate, the Coriolis force leads to a weak coupling between those configurations. The energy eigenstates are again symmetric or antisymmetric combinations of the mirror-image configurations, though in this case—called an Ω-doublet level structure—the splitting between the eigenstates is much smaller than the rotational energy. We’ll meet the Ω-doublet structures again, in some of the experiments described below.
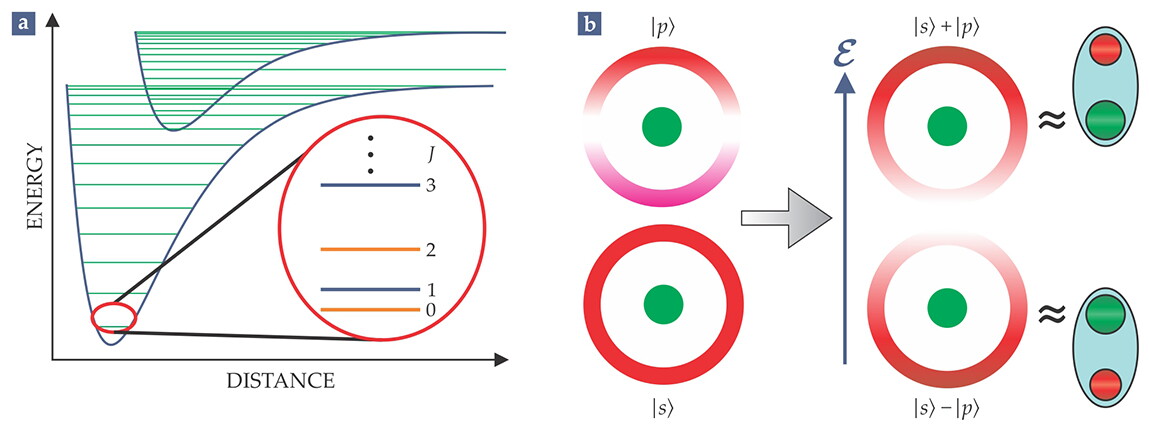
The existence of close-lying energy levels in molecules is ultimately responsible for the molecules’ enhanced sensitivity to tiny perturbations. As one learns in undergraduate quantum mechanics, the effect of a perturbation coupling two states is amplified when the energy difference between the states is small. Figure 2 illustrates a simple and relevant example: The polarizability of a molecule in an electric field is many orders of magnitude larger than that of an atom.

Figure 2. Closely spaced energy levels magnify perturbations. (a) This diagram shows the typical hierarchy of energies in molecules. Blue curves show electronic energies as a function of the distance between atoms in the molecule. Green lines show ladders of vibrational energies. The lowest states of a diatomic molecule have quantized rotational angular momentum and alternating parity, as shown in the blowup. Here, J specifies the rotational angular momentum in units of ℏ. (b) A diatomic molecule can be almost completely polarized by a relatively modest electric field. Wavefunctions for rotational motion around the center of mass are analogous to the familiar |s⟩, |p⟩, and other orbitals of an electron in a hydrogen atom. With that in mind, let |s⟩ denote the ground-state, parity-even, J = 0 wavefunction and |p⟩ the parity-odd, first-excited-state, J = 1 wavefunction. The illustration to the left shows those wavefunctions for a light, negatively charged atom (red) orbiting a heavy positive atom (green); shading of the red wavefunction denotes amplitude, with pink negative and white zero. An electric field ℰ mixes opposite-parity levels. Assuming that it’s OK to consider only the two states with lowest energies Es and Ep, the perturbed wavefunctions are |s̃⟩ ∝ |s⟩ + η|p⟩ and |p̃⟩ ∝ |p⟩ − η|s⟩. Constructive and destructive interferences yield configurations in which the molecule polarizes along the field (|s̃⟩) or against it (|p̃⟩). For weak fields, η ∝ dℰ/(Ep − Es), where d is the electric dipole moment of the molecule. If, however, the electric field is strong enough that dE ≳ Ep − Es, η approaches 1, and the molecular polarization is nearly complete, as illustrated on the right.
CP violation
Several experiments are using strongly electrically polarized molecules in searches for a tiny EDM along the spin of the electron. The electron EDM (eEDM), much like its anomalous magnetic moment (described beautifully in a Physics Today Quick Study by Gerald Gabrielse, December 2013, page 64 ), can arise from a coupling of the electron to virtual particles in loops of Feynman diagrams such as shown in figure 3. However, the eEDM has a special property: It violates time-reversal (T) invariance, and so, according to the CPT theorem of quantum field theory, it also violates CP symmetry. Hence, an eEDM can occur only in the presence of virtual particles that are themselves subject to CP-violating interactions. The standard model of particle physics predicts an extremely small, nonvanishing eEDM, but many proposed extensions to the standard model include new particles and forces that could induce a much larger value. The sensitivity of recent experiments to the eEDM is sufficient to place very strong bounds on many phenomena underlying standard model extensions. 3

Figure 3. Searching for the electron’s electric dipole moment (eEDM). (a) In quantum field theories, the electron continuously emits and reabsorbs virtual particles of all types that exist in nature. An eEDM arises if the charge distribution of that cloud of virtual particles is not spherically symmetric. Shown here is a typical process that could generate an eEDM: The electron splits into a virtual pair of supersymmetric particles—the photino γ̃ and the selectron ẽ—that are partners of ordinary particles. The required violation of time-reversal invariance, denoted by an “x” in the figure, comes from the mechanism that causes supersymmetric particles’ partners to be more massive than their ordinary counterparts. (b) The resulting eEDM d, depicted here as a small asymmetry of charge along the electron’s spin axis, can interact with the internal electric field in a molecule. (c) In the ACME experiment described in the text, a putative eEDM interacts with a strong internal electric field ℰint in a polarized thorium monoxide molecule. That interaction shifts the energy from that of the black line to that of the red. In the ACME experiment, we flip the relative orientations of d and ℰint and measure the double energy shift indicated by the green, double-headed arrows. The oppositely directed shifts for oppositely polarized states help us suppress systematic errors.

The main advantage of the molecules is that the electrons in them can experience an enormous intramolecular electric field ℰint as large as about 100 GV/cm. In the presence of an eEDM, that huge field creates a measurable energy shift. Not surprisingly, ℰint is proportional to the electric polarization of the molecule. Due to relativistic effects, it also rapidly increases with the atomic number of the heaviest atom in the molecule. The sensitivity to an eEDM increases proportionally with the time over which the internal field acts, whereas the statistical measurement precision grows as the square root of the number of molecules observed.
Last year the ACME (Advanced Cold Molecule Electron EDM) collaboration, which I colead along with Harvard University’s John Doyle and Gerald Gabrielse, reported the results of a search for the eEDM using thorium monoxide molecules (see reference and Physics Today, April 2014, page 15 ). We chose ThO because it has an exceptionally large ℰint and because its Ω-doublet level structure makes it easy to fully polarize parallel or antiparallel to a modest laboratory electric field.

Students from Yale and Harvard Universities work on the ACME experiment, a search for the electron’s electric dipole moment. (Photo by Adam West.)

The ACME experiment relied on a new method, developed by Doyle’s group, that used a cryogenic buffer and carrier gas to produce molecular beams with high flux and slow velocity. As a result we were able to obtain unprecedented sensitivity. The eEDM reported by ACME was consistent with zero. Moreover, ACME determined that the magnitude of the eEDM is less than 9 × 10−29 e·cm, a value so small as to rule out most models with CP-violating forces arising from exchange of particles with masses in the few-TeV range—the energy scale currently being explored directly at the Large Hadron Collider at CERN. Future planned experiments should allow ACME to probe energies well beyond those directly accessible at the collider.
Violation of parity symmetry
The electroweak interactions violate spatial-inversion symmetry. From the 1970s to early in this century, atomic-physics experiments looking for parity violation focused mainly on a particular effect known as the weak charge of the nucleus. In the course of those investigations, experimenters also observed a much smaller parity-violating effect that arises from the so-called anapole moment of the nucleus. 5 The anapole moment describes a distribution of current, illustrated in figure 4, that corresponds to a toroidal winding whose central axis parallels the spin of the nucleus. That current distribution can arise due to electroweak forces within the nucleus that cause the spin direction of an individual nucleon to tip in the direction of its momentum.

Figure 4. Bringing energy levels together to reveal an anapole moment. (a) The lower part of this Feynman diagram shows electroweak interactions between the nucleons in a nucleus, mediated by the exchange of virtual W and Z bosons. (b) Those interactions lead to a toroidal current circulating around the nucleus. The central axis of the torus and the spin I of the nucleus are parallel. The electron spin interacts magnetically with the field in the torus, as depicted in panel a by the exchange of a virtual photon. (c) The Zeeman effect is the splitting of degenerate energy levels by an applied magnetic field. By using that effect to tune opposite-parity rotational levels (purple and orange) to near degeneracy, experimenters dramatically enhance the degree of parity violation arising from the anapole moment of a nucleus in the molecule. Typically the field needed to achieve that near degeneracy is about 1 T. In the kets to the right, the large arrows denote electron spins responsible for the Zeeman shift, and the small arrows represent the nuclear spins responsible for the anapole moment.
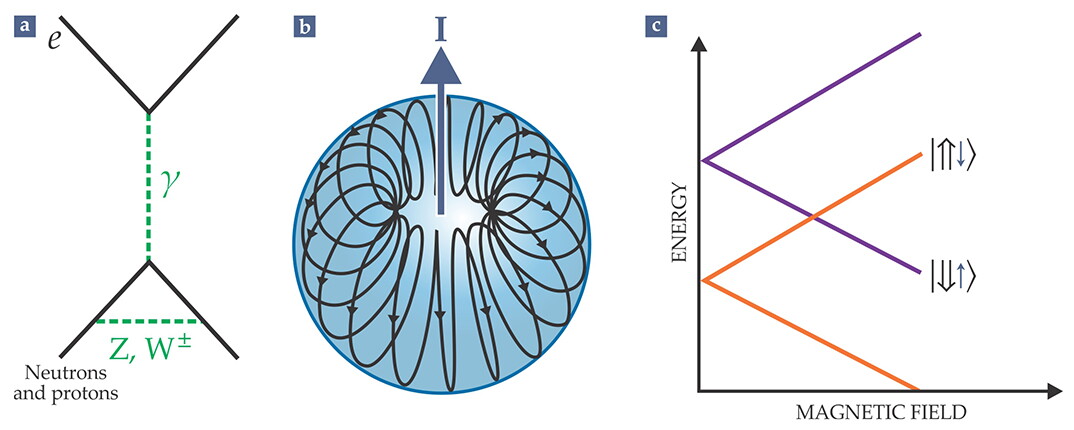
A toroidal winding creates a magnetic field only inside the torus, so the effect of the anapole moment is confined to the nucleus. However, the electron in an atom or molecule spends some of its time inside the nuclear volume. Hence the electron’s spin magnetic moment interacts with the parity-violating anapole magnetic field.
In atoms, that interaction leads to mixing of opposite-parity energy levels that typically are separated by something like 1 eV. In molecules, electronic interactions with the nuclear anapole field can mix opposite-parity rotational levels separated by a mere 10−4 eV or so. In fact, the rotational levels are so close that, thanks to the Zeeman effect, their energy difference can be completely bridged 1 , 6 by applying a magnetic field of order 1 T. Under those conditions, the effective energy separation between the molecular states is determined by a finite energy resolution, which by the uncertainty principle is inversely proportional to observation time.
In 2014 my research group succeeded in bringing opposite parity levels to within about 10−11 eV of each other in barium monofluoride. 6 Such tuning enhances parity-violation-induced mixing by 11 orders of magnitude relative to that in atoms, and we anticipate it will enable measurements of anapole moments for many nuclei. The cesium-atom measurement of the anapole moment—the only observation on the books at present—is difficult to reconcile with data from parity-violating nuclear scattering experiments. A systematic program of molecular measurements should help to clarify how the strong nuclear force modifies electroweak interactions in general, and the anapole-moment-inducing interactions in particular.
Constants that change?
Physicists have long speculated that quantities thought of as constants of nature might in fact vary slightly over time. 7 To test that idea, one might identify pairs of atomic or molecular levels whose energy splittings depend differently on fundamental constants. Then, if the precisely measured ratio of the energy splittings were to change over time, that would be evidence that the dimensionless constants determining the splittings were also varying.
Molecular rovibrational energies depend on the ratio μ of the proton mass to the electron mass, and the splittings that arise due to tunneling are exponentially sensitive to that parameter. Comparing such a sensitive splitting to one that is less sensitive—say, from electronic or vibrational motion—can serve to probe whether μ is varying. Such comparisons can be made directly in the laboratory, where they may be carried out with extremely high precision—but only over the time scale of a few years. Alternatively, they may be conducted indirectly, by comparing molecular transition energies observed in astrophysical data from distant sources to those measured in the laboratory. The indirect approach is much less precise, but it can span time scales approaching the lifetime of the universe.
The structure of some molecules leads to a dramatic amplification of the effects manifested from a change in μ. 8 The key idea is related to the inspiration behind the Vernier caliper: If the energy of a state associated with one type of motion (say, vibrational or tunneling) happens to be almost identical to that of a state associated with another motion (say, electronic or rotational), then the relative difference in energy between the two accidentally degenerate states can be extraordinarily sensitive to changes in μ.
Indeed, the most sensitive test to date for a variation of μ uses that principle. In the methanol (CH3OH) molecule, some rotational states are very close in energy to states, similar to the HOOH states of figure 1, that are associated with torsional tunneling. Transitions between those close-lying states, and between pairs of ordinary rotational states, have been observed both in extragalactic sources and in the laboratory. 9 A comparison of laboratory and astrophysical data has established that during the past 7 × 109 years, Δμ/μ has been less than 1 × 10−7—an extraordinarily precise limit. Similar experiments based on molecular clouds located in dense regions of the Milky Way have set strong limits on any possible density dependence of μ, as could occur in so-called chameleon-field models of dark energy. 9
Ultracold and trapped molecules
The dense and complex energy spectrum that enhances molecules’ intrinsic sensitivity to effects of fundamental interest also makes molecules difficult to work with in experiments. For example, according to the Boltzmann distribution, at room temperature most molecules have hundreds of quantum states populated. If experiments relying on any individual state are not to be plagued by poor statistics, they must be conducted at low temperatures. In addition, energy resolution scales inversely with measurement time, so the energies of molecules moving more slowly can be measured more precisely. If the motion is slow enough to allow the molecules to be trapped, then the measurement time can be further extended—by orders of magnitude.
So far, all the laboratory measurements probing fundamental physics as described above have come from experiments that used molecular beams with internal temperatures of a few kelvin. However, techniques for producing molecular gases at much lower temperatures have begun to work effectively, and they promise to soon yield dramatic improvements in experiments that use molecules to probe fundamental physical laws.
The most advanced methods for producing ultracold molecular gases rely on first cooling individual atoms, then binding pairs of those atoms to make diatomic molecules. 2 The newly formed molecules inherit the very low center-of-mass velocity of the constituent atoms and can be readily trapped—for example, in an optical lattice. In addition, if the starting point is a low-entropy state such as a degenerate quantum gas, the atoms can be transferred very efficiently to a single quantum state of their internal motion.
For example, Tanya Zelevinsky’s group at Columbia University is developing a laboratory experiment to search for temporal variations in μ by measuring vibrational energies in ultracold, trapped diatomic strontium molecules; the combination of long interaction times and techniques to eliminate systematic errors in the energy measurement promises excellent sensitivity. 10 (The error-reducing techniques are similar to those used in optical atomic clocks, as discussed in Physics Today, March 2014, page 12 .) In diatomic cesium and other molecules, accidental degeneracies exist between states of high vibrational energy and metastable electronic states. Measurements of transition energies between those nearly degenerate states would be extraordinarily sensitive to changes in μ. 8 Shin Inouye’s group at Osaka City University in Japan is undertaking measurements of that type with potassium–rubidium molecules. Such experiments, based on the Vernier principle, can potentially surpass the impressive sensitivity already achieved to variations of μ–but with laboratory measurements only, rather than in combination with astrophysical data.
Cooling molecules with lasers
Nowadays, methods of laser cooling and trapping are being applied directly to molecules. They are providing species distinct from those that can be assembled out of laser-cooled atoms and that have specific structures optimized for particular experimental goals. That development is remarkable, as it was long believed that the complex structure of molecules made them essentially impossible to cool with lasers.
Laser cooling relies on the scattering of something like 100 000 photons, because each photon can carry away only a small amount of momentum. In molecules, the absorption of an optical photon excites an electronic orbital, which can then decay to the ground-state orbital. That decay, however, typically excites vibrational or rotational motion. Since each photon carries one unit of angular momentum, the rotational quantum number cannot change by more than 1.
Vibrational excitations are a different story, however. When the electron orbital de-excites, the number of vibrational quanta excited is determined by Franck–Condon factors—overlaps of excited- and ground-state vibrational wavefunctions—that follow no simple rules. In a typical molecule, a single scattered photon can excite more than a dozen different vibrational states with significant probability. Once the molecule is vibrationally excited, the frequency of the laser photons no longer matches the new resonance condition needed to excite the electron’s orbital. So the only way to keep scattering photons is to add many new laser frequencies to keep exciting the molecule from the many populated vibrational states. The repeated process of excitation and de-excitation, called optical cycling, has been accomplished with broadband lasers that hit many molecular resonances at once and also with arrays of single-frequency lasers.
Roughly a decade ago, a pair of seminal papers showed that for a broad class of diatomic molecules, Franck–Condon factors and angular-momentum selection rules are sufficiently simple that just a few single-frequency lasers suffice to maintain optical cycling long enough for laser cooling and trapping. 11 Over the past few years, my group and several others have begun to put those ideas to work. 12 For instance, my group laser cooled strontium monofluoride molecules and demonstrated that their spread of velocities along one direction corresponded to a temperature as low as 300 µK, comparable to that typical of atomic laser cooling. We have also loaded molecules into a magneto-optical trap (MOT). 13 (Figure 5 further illustrates those two experiments.)

Figure 5. Laser cooling and magneto-optical trapping of strontium monofluoride. (a) If a molecule can be induced to scatter enough photons, it can be cooled below a millikelvin. But to have multiple scatterings, an excitation–relaxation sequence must be repeated over and over, a process called optical cycling. Shown here are electronic energy levels and associated vibrational levels (ν for the ground-state ladder and ν’ for the excited-state ladder) relevant for the optical cycling process my colleagues and I used to cool SrF.

The workhorse of atomic laser cooling and trapping, the MOT simultaneously cools and confines; thus it makes possible the accumulation of large, dense samples of ultracold gas. It is the starting point for a vast range of experiments in modern atomic physics involving quantum-degenerate gases, atomic clocks, and more. The ability to apply MOT technology to molecules promises to enable a similarly broad range of new experiments.
The internal, rovibrational motion of molecules can also be cooled and the molecular population thus concentrated in a small number of quantum states. For example, by shaping the spectral profile of a broadband laser so that all vibrational states except the lowest were resonant with some frequency of the light, Pierre Pillet and colleagues were able to drive an initially broad distribution of vibrational levels down to the lowest state. 14 Single-frequency lasers can systematically reduce rotational energy by driving transitions from states with angular momentum Jℏ to states with angular momentum (J − 1)ℏ. The ACME collaboration, for example, used that rotational-cooling technique to enhance the signal size in its eEDM experiment. 4 In contrast to center-of-mass cooling, rovibrational cooling requires only a handful of scattered photons to be effective.
Molecular laser cooling and trapping is still in its infancy, but several groups are making plans to incorporate it into next-generation experiments that probe fundamental interactions. 15 For example, a group of researchers at Imperial College London has begun work on an eEDM experiment in which they plan to launch laser-cooled ytterbium monofluoride molecules in a fountain-like configuration; that design should yield interaction times 100-fold larger than we achieved in our ACME experiment. Larry Hunter’s group at Amherst College and I have together proposed to employ laser cooling (and potentially trapping) of thallium monofluoride molecules in an experiment designed to search for hadronic CP violation via the nuclear “Schiff moment,” which is similar to a nuclear EDM. A group at Groningen University is planning an experiment to measure the anapole moment of the 87Sr nucleus with laser-cooled and trapped SrF molecules.
Beyond the simplest molecules
In this article I have emphasized advances obtained with neutral diatomic molecules, but closely related work has branched into directions that until recently seemed even more exotic. For example, several groups have developed methods for trapping, cooling, manipulating, and precisely measuring properties of molecular ions. In one such experiment, designed to search for the eEDM, a group at JILA has demonstrated extraordinarily long interaction times that promise extremely good energy resolution, even with minimal cooling. 16 A team at the Max Planck Institute of Quantum Optics showed they could trap and laser cool polyatomic molecules. 17 That method might provide a tool for measuring energy levels in complex molecules with unprecedented precision. A long-term goal would be to demonstrate the long-predicted difference in energy between left- and right-handed enantiomers of chiral molecules due to the electroweak interaction. 18
Precision experiments with ultracold, trapped molecules appear set to make a vital contribution toward exploring the frontiers of fundamental physics for years to come.
Box. Estimates of vibrational and rotational energy
The typical energy scale for the motion of electrons in atoms or molecules can be derived beginning from the quantization of angular momentum mvr ~ ℏ for an electron with mass m and velocity v orbiting at a distance r. (Read “~” as “is on the order of.”) The centripetal acceleration from the Coulomb force acting on an electron with charge −e is mv2/r ~ e2/r2. Together, the angular-momentum and acceleration conditions imply that the electron orbits at roughly the Bohr radius a0 = ℏ2/me2 and that the electronic energy Eel ~ e2/r ~ mv2 ~ me4/ℏ2.
In a molecule, atoms with reduced mass M, typically many times the proton mass, are bound together with a binding energy Eb ~ Eel, and each orbits the center of mass at a distance R ~ a0. The energy scale associated with vibrational motion can be understood by modeling the molecule as a harmonic oscillator with spring constant k, so that Eb ~ kR2. The vibrational energy is Evib = ℏ(k/M)1/2 ~ Eel(m/M)1/2; thus Evib/Eel ~ (m/M)1/2 ~ 10−2. The quantized energy of molecular rotation is Erot ~ ℏ2/I, where the moment of inertia I ~ MR2. Hence,
I thank the many colleagues who made possible my contributions to the topics discussed here.
References
1. M. G. Kozlov, L. N. Labzowsky, J. Phys. B 28, 1933 (1995); https://doi.org/10.1088/0953-4075/28/10/008
O. P. Sushkov, V. V. Flambaum, J. Exp. Theor. Phys. 48, 608 (1978);
V. V. Flambaum, I. B. Khriplovich, Phys. Lett. A 110, 121 (1985). https://doi.org/10.1016/0375-9601(85)90756-X2. L. D. Carr et al., New J. Phys. 11, 055049 (2009). https://doi.org/10.1088/1367-2630/11/5/055049
3. S. Barr, Int. J. Mod. Phys. A 08, 209 (1993); https://doi.org/10.1142/S0217751X93000096
M. Pospelov, A. Ritz, Ann. Phys. 318, 119 (2005); https://doi.org/10.1016/j.aop.2005.04.002
J. Engel, M. J. Ramsey-Musolf, U. van Kolck, Prog. Part. Nucl. Phys. 71, 21 (2013). https://doi.org/10.1016/j.ppnp.2013.03.0034. J. Baron et al. (ACME collaboration), Science 343, 269 (2014). https://doi.org/10.1126/science.1248213
5. W. C. Haxton, C. E. Wieman, Annu. Rev. Nucl. Part. Sci. 51, 261 (2001); https://doi.org/10.1146/annurev.nucl.51.101701.132458
V. A. Dzuba, V. V. Flambaum, Int. J. Mod. Phys. E 21, 1230010 (2012). https://doi.org/10.1142/S021830131230010X6. M. G. Kozlov, L. N. Labzovskii, A. O. Mitrushchenkov, J. Exp. Theor. Phys. 73, 415 (1991);
D. DeMille et al., Phys. Rev. Lett. 100, 023003 (2008); https://doi.org/10.1103/PhysRevLett.100.023003
S. B. Cahn et al., Phys. Rev. Lett. 112, 163002 (2014). https://doi.org/10.1103/PhysRevLett.112.1630027. J.-P. Uzan, Rev. Mod. Phys. 75, 403 (2003); https://doi.org/10.1103/RevModPhys.75.403
V. V. Flambaum, Int. J. Mod. Phys. A 22, 4937 (2007). https://doi.org/10.1142/S0217751X070382938. V. V. Flambaum, M. G. Kozlov, Phys. Rev. Lett. 99, 150801 (2007); https://doi.org/10.1103/PhysRevLett.99.150801
D. DeMille et al., Phys. Rev. Lett. 100, 043202 (2008); https://doi.org/10.1103/PhysRevLett.100.043202
P. Jansen et al., Phys. Rev. Lett. 106, 100801 (2011). https://doi.org/10.1103/PhysRevLett.106.1008019. J. Bagdonaite et al., Science 339, 46 (2013); https://doi.org/10.1126/science.1224898
S. A. Levshakov, M. G. Kozlov, D. Reimers, Astrophys. J. 738, 26 (2011), https://doi.org/10.1088/0004-637X/738/1/26 ;
S. Truppe et al., Nat. Commun. 4, 2600 (2013).10. T. Zelevinsky, S. Kotochigova, J. Ye, Phys. Rev. Lett. 100, 043201 (2008). https://doi.org/10.1103/PhysRevLett.100.043201
11. M. D. DiRosa, Eur. Phys. J. D 31, 395 (2004); https://doi.org/10.1140/epjd/e2004-00167-2
B. K. Stuhl et al., Phys. Rev. Lett. 101, 243002 (2008). https://doi.org/10.1103/PhysRevLett.101.24300212. E. S. Shuman, J. F. Barry, D. DeMille, Nature 467, 820 (2010); https://doi.org/10.1038/nature09443
M. T. Hummon et al., Phys. Rev. Lett. 110, 143001 (2013); https://doi.org/10.1103/PhysRevLett.110.143001
V. Zhelyazkova et al., Phys. Rev. A 89, 053416 (2014). https://doi.org/10.1103/PhysRevA.89.05341613. J. F. Barry et al., Nature 512, 286 (2014). https://doi.org/10.1038/nature13634
14. M. Vitaeu et al., Science 321, 232 (2008). https://doi.org/10.1126/science.1159496
15. M. R. Tarbutt et al., New J. Phys. 15, 053034 (2013); https://doi.org/10.1088/1367-2630/15/5/053034
L. R. Hunter et al., Phys. Rev. A 85, 012511 (2012); https://doi.org/10.1103/PhysRevA.85.012511
J. E. van den Berg et al., J. Mol. Spectrosc. 300, 22 (2014). https://doi.org/10.1016/j.jms.2014.02.00416. H. Loh et al., Science 342, 1220 (2013). https://doi.org/10.1126/science.1243683
17. M. Zeppenfeld et al., Nature 491, 570 (2012). https://doi.org/10.1038/nature11595
18. M. Quack, J. Stohner, M. Willeke, Annu. Rev. Phys. Chem. 59, 741 (2008). https://doi.org/10.1146/annurev.physchem.58.032806.104511
More about the authors
Dave DeMille (david.demille@yale.edu ) is a professor of physics at Yale University in New Haven, Connecticut.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}