Designing clusters for heterogeneous catalysis
DOI: 10.1063/PT.3.4248



In 1987 Masatake Haruta and his colleagues at the Osaka National Research Institute in Japan reported on catalytic behavior of gold nanoclusters. 1 Theirs was not the first work to demonstrate metal nanoclusters as catalysts, but that discovery was striking given bulk gold’s well-known chemical inertness. The startling change in the behavior of gold when its size was reduced drew renewed attention to the study of metal nanocluster catalysts.
Today catalysis is involved at some point in more than 90% of all chemical manufacturing processes. Most catalysts used for those and other large-scale processes—including fuel conversion and abatement of waste from vehicles and power plants—are porous, high-area solids with nanoparticles dispersed on the internal surfaces. Such catalysis is complex because it occurs on surfaces that are heterogeneous in both composition and structure. Thus technological advances are often the result of trial-and-error discovery. The dream for many catalysis researchers is first-principles design of catalysts with optimized properties, such as high efficiency, stable long-term operation, and regeneration after degradation caused by processes such as sintering and undesired side reactions, whose products block the surface.
The metals used in catalysts are often scarce and expensive, and only the metal atoms on the surface do catalysis. Subnanometer particles, called clusters, that consist of just a few atoms increase the surface area per metal atom. Figure
Figure 1.

Various forms of platinum catalysts. From left to right are bulk Pt(111), a Pt nanoparticle consisting of 146 atoms supported on magnesium oxide (100), and a seven-atom Pt cluster on MgO(100). In the cluster, all the Pt atoms are exposed and interact with reactants.

Manufacturers already use metal clusters as industrial catalysts—for example, platinum clusters on the order of 1 nm in diameter convert hydrocarbons, such as n-hexane and n-heptane, into aromatics, such as benzene and toluene. Surface-supported clusters of a few atoms and isolated atoms of platinum are also present in catalysis for the dehydrogenation of propane, a process that is one route to convert a component of shale gas into polymers and other chemicals.
The structures of those catalysts must be highly stable to be useful in industry. But metal nanoclusters on surfaces tend to coalesce into larger nanoparticles and lose catalytic activity as they lose surface area. They may be stabilized by metal–support interactions, as we discuss later. The reactions with metal clusters are different from those with the bulk metals, and they depend on the cluster size. 2
Cluster reactivity
Manufacturers produce industrial catalysts by methods that are large scale and cost effective but imprecise. For example, an aqueous solution of tetraammineplatinum(II) nitrate, Pt(NH3)4(NO3)2, placed in contact with a porous metal oxide gives absorbed platinum salts that, when heated in air and then treated in hydrogen, are converted into platinum clusters and nanoparticles of various sizes, including isolated atoms. Because the resulting clusters and nanoparticles are not uniform, they are not ideal for investigations aimed at understanding how structure affects performance.
Researchers rely on synthesis methods that consistently produce clusters of the same size. One method uses a beam of vaporized metal that is chosen for a particular size of its gas-phase clusters. When the vapor hits a nonporous planar support, it deposits clusters; some are the same size as the gas-phase clusters, and some break apart on impact or coalesce. Alternatively, clusters stabilized with ligands—for example, Os3(CO)12 or Ir4(CO)12—react with metal oxide surfaces to produce supported clusters with some intact ligands that may be modified or removed. Although such methods are too expensive for practical application, they have opened the way to vibrant scientific literature that engages both experimentalists and theorists.
Researchers can determine the number of metal atoms per cluster using recent advances in scanning transmission electron microscopy that allow for atomic-resolution images of supported clusters when there is sufficient contrast—that is, for heavy metal atoms on supports consisting of light atoms. For example, images of triosmium clusters on crystalline magnesium oxide, in the top row of figure
Figure 2.

Scanning transmission electron microscopy images and crystallographic models (left) of three-, four-, and five-atom osmium clusters on magnesium oxide. (Adapted from ref.

Even with the best-made samples and a full complement of tools, essential properties of supported metal clusters are beyond experimental reach. Computational modeling provides an in-depth picture of the properties of a polyatomic system and thus helps elucidate the relationship between structure and catalytic properties. Synergy between theory and experiment now drives the development of the field. Nevertheless, as we will discuss later, there are still important limitations of theory and experiment.
Experimental results show that the addition or removal of just one atom can markedly influence the activity, or rate of catalysis, and the selectivity, or ratio of desired to undesired products. For example, theory and experiment jointly showed 5 that Pt7 dispersed on aluminum oxide is significantly more active for alkane dehydrogenation than Pt8. Although the most stable forms of both supported Pt7 and Pt8 have three-dimensional morphologies with limited activities for dehydrogenation of alkanes, Pt7 clusters have easy thermal access to metastable quasi-2D isomers that are significantly more active. Those active species actually do the catalysis. In contrast, the most stable 3D structure of Pt8 clusters is dominant at all temperatures, and Pt8 is overall less active. When supported on rutile (TiO2), however, Pt8 is a more active catalyst for CO oxidation than smaller platinum clusters. 6 Minor changes in cluster size, interactions with the support, and composition lead to major changes in catalytic properties, which open the door to catalyst tuning.
The support surface affects the nanoscale catalysts on it because most of the metal atoms are at the interface and interact with the surface underneath. 7 That interaction alters the morphology of the cluster, and it has a marked influence on the catalyst performance. Some supports, such as TiO2, CeO2, and other reducible oxides, tend to withdraw electrons from the metal, whereas some nonreducible oxides, including MgO and Al2O3, donate electrons. Although the magnitude of the charge transfer between the support and metal is usually small, it is sufficient to alter catalyst performance. Moreover, researchers can enhance the support’s influence by preparing clusters nestled in support-surface defects such as metal or oxygen vacancies, which are common in oxides.
Fluxional and polymorphous
Until recently theorists working with metal clusters used simplified models that treated clusters as almost static entities. For a given size and composition, scientists determined the most stable structure, the ground state, and the chemical and catalytic properties only for that structure. Now theory compares the properties and structures of various isomers that are present during catalysis with similar energies but different catalytic properties. Notably, the most stable isomer may not be the most catalytically active for the desired reaction.
Let us consider Pt7 supported on a perfect MgO(100) surface. Figure
Figure 3.

The seven-atom platinum isomers, shown from above, that are theoretically predicted to be energetically accessible on a perfect magnesium oxide (100) surface at 700 K. The isomers’ different shapes and different bonding affinities influence their catalytic properties.

Theorists need better electronic-structure methods to overcome some of the limitations of DFT. For many structural forms of clusters, the problems average out, and the calculated structures qualitatively agree with experimental observations. But a main weakness of DFT is its inability to qualitatively capture the behavior of strongly correlated systems, such as manganese, iron, and cobalt oxides and sulfides.
Beyond the static structure, clusters convert from one isomer to another as the adsorbed molecules and reaction intermediates change over the course of a catalytic cycle. Structural changes are possible because the chemical bonds are delocalized and nondirectional in metal clusters. That dynamic structure, or fluxionality, is pronounced in catalytic processes because the temperatures are high, typically 700–800 K. The support also facilitates or hinders certain morphologies. But cluster fluxionality is not in reach of today’s experimental capabilities. Researchers would need operando measurements capable of targeting one particular cluster on extremely short time scales and in complex environments. For now, theory and computation are leading the way toward understanding the fluxional character of the cluster catalysts.
One of our papers introduced new DFT-based algorithms that enabled investigations of the fluxional character, including the probability the shape will change and the time scales associated with those changes.
8
But the computational cost for a system of only seven Pt atoms and the support surface is still enormous. Extensions to more complex catalysts will require further improvements in computational capabilities and algorithm efficiency. Nonetheless, the available results already show that dozens of possible surface-supported Pt7 isomers exist under the conditions of catalysis, as shown in figure
Figure 4.

The interconversion of seven-atom platinum isomers on α-alumina indicates that the clusters are divided into two groups (circled in red and green). Within each group, the probability for cluster isomerization is high because energy barriers are low. But between groups, the probability is lower because the energy barriers are high. The most stable isomer (labeled as 1) belongs to the group circled in red, but that isomer is catalytically inactive. The second most stable isomer (labeled as 2) belongs to the second group (in green), and it is highly active catalytically. (Adapted from ref.

The number of thermally accessible isomers varies substantially and depends on the nature of the support, the number of atoms in the cluster, the cluster’s stoichiometry, and its coverage with reactants, intermediates, and products. The complexity arises from the balance of forces from the intracluster electronic structure, cluster–support bonds, and cluster–molecule bonds. But that balance is not yet predictable. Only massive stochastic simulations can account for all of those effects.
Understanding the dynamics and fluxionality of supported clusters is a significant step forward, but that understanding alone can’t predict the structures in practical catalysis without considering realistic conditions. In practice, the cluster shape depends on other aspects of the surrounding environment, including the structure of the supports, such as the nanopores of zeolites that envelop metal clusters, and the steady-state distribution of the bound reactants, intermediates, products, and impurities. Theorists still have a long way to go before real-world catalysts can be designed and understood from first principles. 9
Interactions with adsorbates
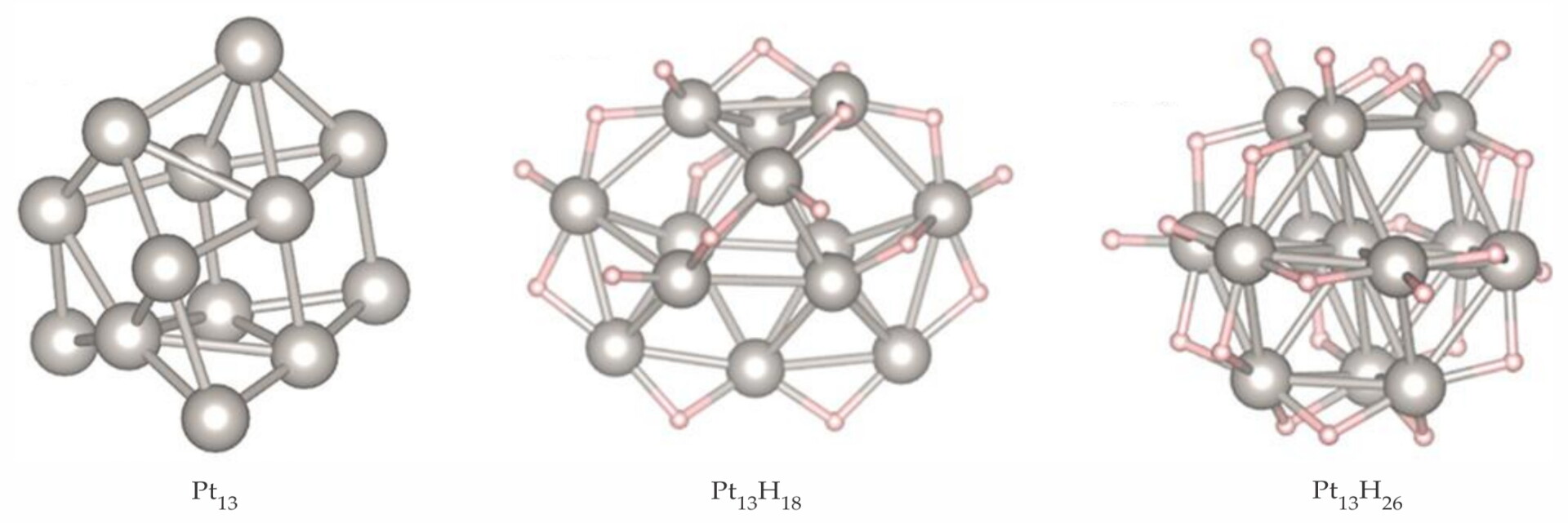
One step toward realistic catalyst models is accounting for adsorbates on the clusters. They vary during a catalytic cycle—reactants on, intermediates formed, products off—and can dramatically modify the size and shape of cluster isomers. 10 For Pt13 clusters in an atmosphere of hydrogen, common in dehydrogenation catalysis, Geng Sun and Philippe Sautet of UCLA used a simplified model that ignored the influence of the support, because high coverage of hydrogen causes the particle to become globular and lose some interactions with the support. They found that the adsorbate exerts a marked effect on the Pt13 cluster morphology. 11
Without adsorbates, many isomers are energetically accessible at high temperatures. Using methods similar to those for Pt7 clusters, Sun and Sautet predict that the most stable geometry of Pt13 is a tricapped pentagonal prism, shown in the left panel of figure
Figure 5.

The most stable 13-atom platinum clusters with increasing adsorbates are shown for no hydrogen (left panel), 18 H atoms (center), and 26 H atoms (right). The cluster shape is transformed under the influence of a hydrogen atmosphere. (Adapted from ref.
The daunting complexity of real catalysis
Real supported metal cluster catalysts are complex mixtures, with their properties influenced by the cluster size, the support, and the adsorbates, all of which influence cluster morphology. Accurate predictions of the catalytic properties would require a tour de force of modeling or incisive operando characterization by imaging and spectroscopy, both far beyond our current capabilities.
Theoretical demonstrations show that clusters’ electronic structures, morphologies, and interactions with supports strongly influence catalytic performance and that cluster fluxionality is important. Those results help experimentalists realize the limitations of data that characterize catalysts that are not in the working state; they also show the importance of developing high-speed imaging techniques that are orders of magnitude faster than today’s capabilities to capture rapid morphological changes, which occur in tens of picoseconds. The insights emerging from both theoretical and experimental research are beginning to guide practical catalyst discovery—for example, by focusing development on small clusters, clusters that are stabilized by their interactions with supports, and supports that optimize those interactions.

Platinum cluster catalysts on magnesium oxide.

References
1. M. Haruta et al., Chem. Lett. 16, 405 (1987). https://doi.org/10.1246/cl.1987.405
2. Z. Xu et al., Nature 372, 346 (1994). https://doi.org/10.1038/372346a0
3. C. Aydin et al., Angew. Chem. Int. Ed. 52, 5262 (2013). https://doi.org/10.1002/anie.201300238
4. A. Kulkarni et al., Angew. Chem. Int. Ed. 49, 10089 (2010). https://doi.org/10.1002/anie.201005105
5. E. T. Baxter et al., ACS Catal. 7, 3322 (2017). https://doi.org/10.1021/acscatal.7b00409
6. Y. Watanabe et al., Catal. Sci. Technol. 1, 1490 (2011). https://doi.org/10.1039/c1cy00204j
7. A. Kulkarni, R. J. Lobo-Lapidus, B. C. Gates, Chem. Commun. 46, 5997 (2010). https://doi.org/10.1039/c002707n
8. H. Zhai, A. N. Alexandrova, J. Phys. Chem. Lett. 9, 1696 (2018). https://doi.org/10.1021/acs.jpclett.8b00379
9. E. Jimenez-Izal, A. N. Alexandrova, Annu. Rev. Phys. Chem. 69, 377 (2018). https://doi.org/10.1146/annurev-physchem-050317-014216
10. A. M. Argo et al., Nature 415, 623 (2002). https://doi.org/10.1038/415623a
11. G. Sun, P. Sautet, J. Am. Chem. Soc. 140, 2812 (2018). https://doi.org/10.1021/jacs.7b11239
More about the authors
Elisa Jimenez-Izal is an Ikerbasque research fellow at the University of the Basque Country and the Donostia International Physics Center, both in Donostia, Spain. Bruce Gates is a professor of chemical engineering at the University of California, Davis. Anastassia Alexandrova is a professor of chemistry and biochemistry at UCLA and the California NanoSystems Institute there.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}