Beyond quantum jumps: Blinking nanoscale light emitters
DOI: 10.1063/1.3086100
Imagine driving your car at night while its headlights display an annoying blinking behavior, switching on and off randomly. To add to the nuisance, the blinking has no definite time scale. In fact, although in most of your nightly journeys your headlights display quite rapid blinking, rendering at least some visibility, occasionally they remain off for almost the entire journey.
Ridiculous and impractical as that behavior may seem, such is the situation commonly encountered by nanoscientists: A wide variety of natural and artificial nanoscopic light emitters, from fluorescent proteins to semiconductor nanostructures, display a blinking behavior like that described above. The emission (on) and no-emission (off) periods have a duration that varies from less than a millisecond to several minutes and more. The probability of occurrence of the on and off times is characterized by a power law, which is a typical sign of high complexity and is fundamentally different from what is expected from the quantum jump mechanism of fluorescence blinking predicted at the dawn of quantum mechanics.
So what is the origin of the power-law blinking? Since it is a nearly universal behavior of single emitters, we tend to think there must be a fundamental answer. Remarkably, even a decade after the behavior was discovered, a satisfactory explanation of the power-law blinking has managed to evade all the experimental and theoretical efforts. That enigmatic luminescence blinking and its peculiar statistical consequences are the focus of this article.
Quantum jumps and fluorescence blinking
Almost a century ago, Niels Bohr proposed his now famous model in which electrons occupy discrete energy levels, or orbits, within the atom. That energy discretization led Bohr to the “quantum jump” prediction: Since electrons cannot be between states, they must undertake instantaneous leaps from one state to another. Direct experimental observation of quantum jumps had to wait until the mid-1980s, when individual ions could be trapped and addressed optically. The jumps were detected as interruptions in the fluorescence emission of single ions when a second electronic transition from a common ground or excited state was pumped in parallel (see
The breakthrough of single-molecule detection brought unprecedented detail into many areas of physics and led to the discovery of numerous surprising effects. Individual nanoscale emitters can be used as fluorescent markers for a number of physical and chemical processes. By attaching them to large biomolecules, for instance, one can directly observe conformational changes and molecular function. Furthermore, one may follow trajectories of individual molecules and thereby track dynamics in a biological cell, for example. The great potential of single-molecule measurements is often restricted by two phenomena. The first one is obviously the blinking: If it occurs on a time scale relevant to the experiment, it complicates the retrieval of information. Second, measurements are limited by photobleaching: Excited molecules may use their excess energy to undergo irreversible chemical reactions that render them nonfluorescent.
Power-law blinking
Studies of the long time scales of blinking events had to await the construction of bleaching-resistant emitters. Those nanoscopic light sources, enabling virtually infinite measurement times, were semiconductor nanocrystals, commonly referred to as colloidal quantum dots. Described in
Fluorescence blinking of individual QDs was first observed in 1996 by a collaboration between the groups of Moungi Bawendi at MIT and Louis Brus, then at Bell Laboratories. 2 Actually, it was surprising to find QD blinking at all because no mechanism known at the time would affect a dot’s emission. Adding to the surprise, the researchers immediately realized that the sojourn times—the times spent in the on and off states—were not exponentially distributed, a result that implied the presence of complex processes behind the new blinking phenomenon. Later, Masaru Kuno, David Nesbitt, and coworkers at JILA found that the probabilities of the on and off times follow a power law (see figure 1) of the form

Figure 1. Fluorescence blinking of colloidal quantum dots. (a) Confocal fluorescence image of individual QDs. (b) Two-minute fragment of a time trace of the emission of a single QD. The blinking can be readily observed. (c) A two-second zoom-in. The horizontal line marks the threshold used to discern the off state from the on state. (d) The distribution of times in the on and off states. The line is a power law (equation
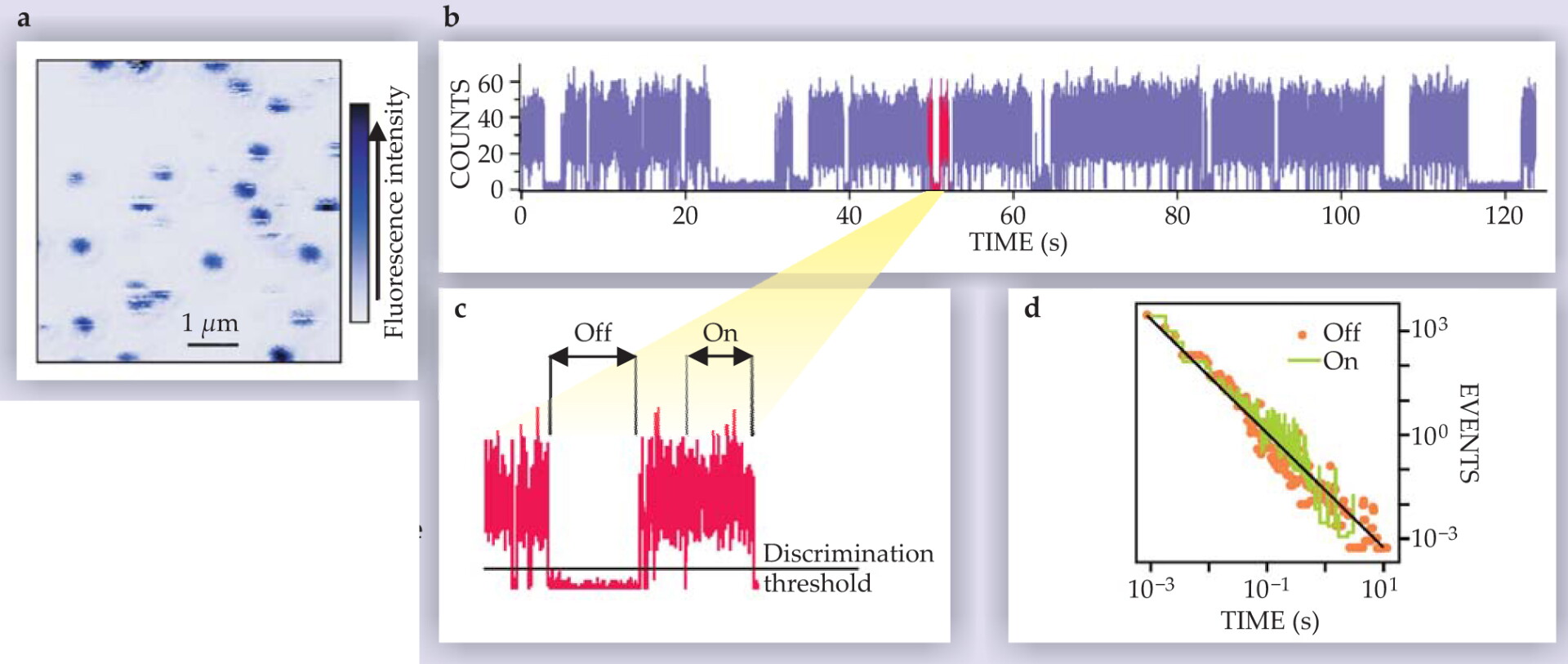
where t is the length of the on or off sojourn time and the exponent α lies between 1 and 2 (typically close to 1.5). 3
The emission of single colloidal QDs can be monitored for hours, and the on and off times have a power-law distribution over several decades in time. Further experimentation showed that all colloidal QDs display the same kind of behavior, regardless of their size, composition, and structure—whether the QD consisted of a bare semiconductor core or of a core surrounded by an inorganic shell. Although an accurate determination of α is not trivial, some studies indicate that the on- and off-state exponents are identical and that they are independent of temperature and of the intensity of the exciting light source. Yet when the intensity is strong enough, exponential cutoffs of the distributions of the sojourn times are observed, usually leading to shorter maximum on times than off times. 4
In recent years different routes to circumvent photobleaching in other systems led to the discovery that power-law blinking is not exclusive to QDs. 5 The same power-law blinking has been uncovered in almost any type of single quantum emitter embedded in a disordered medium, including semiconductor nanorods and longer nanowires, organic molecules, fluorescent proteins, and conjugated polymers.
Quantum jumps in three-level systems
The first observations of quantum jumps were made in the mid-1980s when trapping and optical spectroscopy of individual ions became feasible. Those initial observations used a single ion (Ba+ or Hg +) that can undergo two distinct electronic transitions from one common state: a strong, highly probable transition and a weak, much less frequent one. When the ion was illuminated with light resonant with both transitions, the strong transition dominated and the ion performed many cycles of excitation and de-excitation, emitting a continuous stream of fluorescence photons. Eventually, the much less probable weak transition took place, with a de-excitation time orders of magnitude longer. Thus quantum jumps to the weak level were easily detected because they momentarily interrupted the strong fluorescence emission. In other words, the quantum jumps were evidenced by the fluorescence blinking.
As in the case of the ions, a molecule under appropriate illumination undergoes excitation and de-excitation cycles between singlet states, which leads to a stream of fluorescence photons (the fluorescence is “on”). Eventually, the excited molecule can undergo a transition to the lower-lying triplet state. Transitions between singlet and triplet states are forbidden by symmetry. In practice, they arise with small probability due to spin-orbit coupling. The decay from the triplet state to the singlet ground state therefore takes a relatively long time. The fluorescence emission is interrupted (“off”) during the residence time in the triplet state, so here, too, a blinking fluorescence signal is observed. Such blinking is evident in the time trace. The times spent in the on state (above the horizontal intensity threshold) and in the off state (below the threshold) are exponentially distributed, as shown in the histogram, in perfect agreement with the predictions of the quantum jump theory.

Statistical effects
The main property of power laws is their scale invariance. It can be seen from equation
It is evident from equation
Imagine an experiment in which we collect the light emitted by an ideal QD under continuous excitation. For simplicity assume the sojourn times in the on and off states are identically distributed with a probability given by equation
If an average sojourn time exists, as it does when the distribution of sojourn times is exponential, and we make the measurement time T long enough, we would find the usual ergodic result that Ī = 〈I〉. However, if the time scale of the problem is infinite, as is the case with the power law of equation
Colloidal quantum dots
Colloidal quantum dots (QDs), like the cadmium selenide dot in the transmission electron micrograph below, are isolated, individual semiconductor nanocrystals that are chemically prepared with sizes between 2 and 6 nm. For sizes within that range, a number of properties undergo a transition between molecular and bulk values. As the size of the nanocrystals decreases, the motion of charge carriers is more and more restricted, an effect called quantum confinement. As a result, the smaller QDs have a larger bandgap and discrete energy levels. When illuminated with energies above the bandgap, QDs absorb light and generate an electron–hole pair, called an exciton, which may decay via the emission of a photon. By varying the dot’s size and composition one can tune optical absorption thresholds and emission wavelengths across the visible region of the spectrum, as shown in the photograph of beakers with cadmium telluride QDs under UV illumination and in the plot of emission spectra. The dots in the photo range in size from 2.5 nm on the left to 5 nm on the right. From left to right in the plot, the CdTe dot sizes are 2–6 nm, the CdHgTe dots 3–6 nm, and the HgTe dots 2.5–5 nm.
Due to their small size, colloidal QDs have surfaces characterized by atoms with dangling bonds—shown in the schematic—that can deteriorate a dot’s performance by trapping excited electrons. Organic ligands at the surface have a twofold function: They stabilize the colloids in solution and satisfy some of the dangling bonds. Another approach to satisfy the dangling bonds at the QD surface is the growth of an inorganic shell, usually made of another semiconductor of higher bandgap; such quantum dots are usually called core–shell QDs. (Images courtesy of Andrey Rogach, LMU Munich.)

Physical mechanisms
What could be the physical process behind the fluorescence intermittency? Most of the proposed blinking mechanisms are inspired by work done in the late 1980s by Alexander Ekimov, Alexander Efros, and coworkers at the Ioffe Institute in Saint Petersburg, who examined cadmium sulfide nanocrystals embedded in glass matrices. 7 Those nanocrystals were the first that showed size-dependent quantum effects. In that system, Ekimov and colleagues found that electrons from excited QDs could escape from the dot into long-lived trap states in the glass. Furthermore, they observed that the luminescence of an ensemble of CdS QDs decreased with time under constant illumination. That “photodarkening” was explained by a mechanism called photoassisted Auger ionization, which accounted for all experimental observations: A doubly excited QD could expel an electron out of the dot by using the recombination energy of one of the excitons. The lone hole left in the dot rapidly takes up the energy of subsequently generated excitons and thus provides a fast, non-radiative relaxation pathway and reduced luminescence.
Soon after the observation of luminescence blinking of single QDs, Efros and Mervine Rosen at the US Naval Research Laboratory suggested the first possible explanation based on the above picture: the direct manifestation of the dynamics of photoassisted Auger ionization. 8 An off period starts when an electron is expelled from the QD, and it ends once the electron returns via a tunneling or thermally activated process (figure 2(a)). Within that picture, the blinking sequence on → off → on … corresponds to neutral dot → charged dot → neutral dot…. Although that model would explain why blinking occurs in QDs, it does not predict the power-law distributions of on and off times. Further, the requirement of a doubly excited QD suggests a quadratic dependence on the excitation intensity for the switching-off process, but such a dependence is not observed in the experiments. Still, the physical picture is intuitive and sensible, and different mechanisms for the ionization and neutralization of QDs have been investigated theoretically in efforts to account for the experimental observations. 9

Figure 2. Models for power-law blinking based on charge separation. (a) The charged-dot hypothesis states that a neutral quantum dot is fluorescent and a charged dot is dark. The different models attempt to explain the blinking statistics by proposing mechanisms by which a QD may eject or recover a charge carrier; the carrier is depicted here as an electron, but it could equivalently be a hole. (b) An electron ejected from the QD will diffuse three-dimensionally, but the Coulomb interaction will restrict it to the vicinity of the positively charged QD core. (c) An electron can escape the QD through resonant tunneling when the energies of the QD excited state and a trap (T) match. Random fluctuations in the energy of those levels lead to a power-law distribution of on and off times. (d) Tunneling through a barrier to one of multiple traps gives rise to a power-law distribution of off times through the exponential dependence of tunneling probability on the distance r to the trap.

Probably the simplest possibility is to consider that once the electron leaves the QD (such as by the photoassisted Auger process), it may diffuse in the vicinity of the QD before it returns (figure
Instead of diffusion in space, we may consider a “diffusion” of energy levels as suggested by Bawendi and coworkers at MIT. The idea is that electrons may escape from the dot and return to it via resonant tunneling between an excited state in the QD and a trap state, located outside the QD or at the surface (figure
An alternative tunneling mechanism to explain the power-law blinking was suggested by Kuno and coworkers at JILA and by Michel Orrit’s group at the University of Leiden. Here again, an excited electron in the QD may tunnel to a trap site, presumably located on the surface of the capping shell or in the disordered matrix in the vicinity of the dot (figure
Ergodicity breaking in power-law blinking
To get a sense of how the breakdown of ergodicity comes about in power-law quantum dot (QD) blinking, first consider a situation of two gamblers, Paul and Ken, tossing a coin. Every time the coin shows heads (H), Ken gives a dollar to Paul, and when it shows tails (T), Paul gives Ken a dollar. From Paul’s perspective, he is a winner (W) whenever he has more money than Ken, and he is a loser (L) or even (E) otherwise. The sequence HHHTHTTT thus yields WWWWWWWE. If the players play long enough, our naïve expectation would be that Paul will be the winner for half of the time, Ken the other half, and the even situation will rarely occur. However, that is not what happens. In fact, either Paul or Ken will stay on the winning side for nearly all the time.
The time t it takes Paul to return to the E state is called first-passage time of a one-dimensional random walker and is distributed as P(t) ∝ t −3/2. Thus the distribution of sojourn times in W or L states is similar to that for the sojourn times of blinking QDs with a = 3/2. The total time spent as a winner W or loser L is called the occupation time of that state. The French mathematician Paul Lévy (well known for his generalization of the Gaussian central limit theorem; indeed, stochastic processes with power-law tails do not converge to Gaussian behavior and are often called Lévy processes) found that in the long time limit, the occupation times follow a bimodal distribution called the arcsine distribution. In fact, the situation in which Paul is in the W state exactly half of the time is the least likely, in contrast with our initial expectation.
For the case of the QD emission, the occupation times can be translated into the normalized time-average intensity Ī/I 0, and the arcsine distribution becomes
where 0 < Ī/I 0 < 1; the distribution is plotted at right. The bimodal shape of the distribution reflects the fact that a QD spends most of the time either off or on, making it impossible to obtain a time-averaged intensity and thus leading to ergodicity breaking.
Another method to observe ergodicity breaking in the lab is to consider the photon statistics. The number of photons emitted in time t from a single emitter is n = ∫0 t /(t)dt = Īt. So if Ī is a random variable, with large fluctuations, so will be the fluctuations of the photon counts. A measure of photon statistics is Mandel’s parameter Q = (〈n 2〉 − 〈n〉 2)/〈n〉 − 1, where 〈〉 denotes ensemble averaging. In quantum optics, Q is used to quantify fluctuations of light sources. There are two main regimes in the limit of long measurement time. The sub-Poissonian case Q < 0 is found when anti-bunching effects are important, as in the process of single photon emission. The super-Poissonian Q > 0 bunching regime is also common, found, for example, in the molecular blinking discussed in

More puzzles
Each of the above theoretical frameworks has succeeded in explaining some of the characteristics of power-law blinking, but never all of them. For instance, a considerable number of research groups have reported exponents deviating from the α = 3/2 predicted by the diffusion models. The different exponents, which in some cases are found to depend on the environment, could be accounted for by the electron-tunneling model with a distribution of trap states, which also naturally accounts for the insensitivity of power-law blinking kinetics to temperature. However, that model fails to explain the power-law distribution of on times, which follows rather straightforwardly from the energy-level diffusion model. In addition, some observations can’t be explained satisfactorily by any of the models in their present form. For example, a memory effect has recently been uncovered in the distributions of both the on and the off times: A long on time is followed with higher probability by another long on time. 10 The same happens with successive off times, but curiously it does not happen for consecutive on and off times, which hints that the two processes might be independent.
All in all, some experiments have raised more questions than they answered, and conclusive experiments still have to be done. For example, the dependencies of the blinking on the wavelength and intensity of the pump light source, and the effects of external electric and magnetic fields, still need to be thoroughly explored. Given the generally high complexity of which a power-law distribution is reminiscent, it might well be that a combination of various mechanisms is at play. Additionally, theoretical approaches that do not consider QD charging have been hypothesized, 11 but it remains to be seen whether such alternatives will lead to increased insight.
Despite our lack of understanding of how the power-law behavior actually arises, two approaches to influence the blinking have recently been developed. First, in attempts to functionalize QDs for different applications, various ligand molecules have been used to modify the QD surface, and they turn out to have a strong influence on the photophysical behavior. 12 For example, electron-donating ligands lead to a drastic reduction in the length and frequency of the off times. However, the blinking kinetics do not necessarily change concomitantly; although off times occur less frequently, they still follow a power-law distribution. Thus, while an important step for practical applications has been made in reducing the frequency of unwanted blinking events, it remains to be seen what that approach can bring to our understanding of how the power-law behavior arises.
In a second approach, blinking has been suppressed by synthesizing QDs with a core of CdSe QDs surrounded by thick (5–15 nm) shells of highly crystalline CdS. 13 Such findings add to the notion that the power-law blinking is related to a disordered environment and does not arise in a crystalline medium. In fact, it was already known that so-called self-assembled QDs, which under certain conditions spontaneously form during epitaxial growth of high-quality crystals and have a nearly perfect lattice match with the embedding material, do not show fluorescence blinking.
A nonblinking nanoscopic light emitter will be an important breakthrough for practical applications. That the blinking behavior can be influenced by surface modifications makes us think the first steps to achieve that goal have been made. However, that’s not certain. The origin of power-law blinking remains unknown, and as we have seen here, taking random steps can make us spend very long times on the wrong side of where we want to be. Some things are clear: Both the QD structure and its immediate nano-environment play crucial roles. And from the viewpoint of a physicist, it is fascinating to find such rich behavior in a system as small as a nano-sized single emitter and to be challenged to unravel its mechanism.
The authors thank Robert Silbey for discussions and careful reading of the manuscript. Eli Barkai thanks the Israel Science Foundation and the Massachusetts Institute of Technology for support.
References
1. A. P. Alivisatos, Science 271, 933 (1996). https://doi.org/10.1126/science.271.5251.933
2. M. Nirmal, B. O. Dabbousi, M. G. Bawendi, J. J. Macklin, J. K. Trautman, T. D. Harris, L. E. Brus, Nature 383, 802 (1996). https://doi.org/10.1038/383802a0
3. M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher, D. J. Nesbitt, J. Chem. Phys. 115, 1028 (2001). https://doi.org/10.1063/1.1377883
4. K. T. Shimizu, R. G. Neuhauser, C. A. Leatherdale, S. A. Empedocles, W. K. Woo, M. G. Bawendi, Phys. Rev. B 63, 205316 (2001); https://doi.org/10.1103/PhysRevB.63.205316
F. D. Stefani, W. Knoll, M. Kreiter, X. Zhong, M. Y. Han, Phys. Rev. B 72, 125304 (2005). https://doi.org/10.1103/PhysRevB.72.1253045. J. P. Hoogenboom, E. M. H. P. van Dijk, J. Hernando, N. F. van Hulst, M. F. García-Parajó, Phys. Rev. Lett. 95, 097401 (2005); https://doi.org/10.1103/PhysRevLett.95.097401
R. Zondervan, F. Kulzer, S. B. Orlinskii, M. Orrit, J. Phys. Chem. A 107, 6770 (2003). https://doi.org/10.1021/jp034723r6. X. Brokmann, J. -P. Hermier, G Messin, P. Desbiolles, J. -P. Bouchaud, M. Dahan, Phys. Rev. Lett. 90, 120601 (2003); https://doi.org/10.1103/PhysRevLett.90.120601
G. Margolin, E. Barkai, Phys. Rev. Lett. 94, 080601 (2005). https://doi.org/10.1103/PhysRevLett.94.0806017. A. I. Ekimov, A. L. Efros, Phys. Status Solidi B 150, 627 (1988); https://doi.org/10.1002/pssb.2221500244
D. I. Chepic, A. L. Efros, A. I. Ekimov, M. G. Ivanov, V. A. Kharchenko, I. A. Kudriavtsev, T. V. Yazeva, J. Lumin. 47, 113 (1990). https://doi.org/10.1016/0022-2313(90)90007-X8. A. L. Efros, M. Rosen, Phys. Rev. Lett. 78, 1110 (1997). https://doi.org/10.1103/PhysRevLett.78.1110
9. P. Frantsuzov, M. Kuno, B. Jankó, R. A. Marcus, Nat. Phys. 4, 519 (2008); https://doi.org/10.1038/nphys1001
F. Cichos, C. von Borczyskowski, M. Orrit, Curr. Opin. Colloid Interface Sci. 12, 272 (2007); https://doi.org/10.1016/j.cocis.2007.07.012
G. Margolin, V. Protasenko, M. Kuno, E. Barkai, Adv. Chem. Phys. 133, 327 (2006).10. F. D. Stefani, X. Zhong, W. Knoll, M. Han, M. Kreiter, New J. Phys. 7, 197 (2005); https://doi.org/10.1088/1367-2630/7/1/197
J. P. Hoogenboom, J. Hernando, M. F. García-Parajó, N. F. van Hulst, J. Phys. Chem. C 112, 3417 (2008). https://doi.org/10.1021/jp710100b11. P. A. Frantsuzov, R. A. Marcus, Phys. Rev. B 72, 155321 (2005). https://doi.org/10.1103/PhysRevB.72.155321
12. V. Fomenko, D. J. Nesbitt, Nano Lett. 8, 287 (2008). https://doi.org/10.1021/nl0726609
13. B. Mahler, P. Spinicelli, S. Buil, X. Quelin, J.-P. Hermier, B. Dubertret, Nat. Mater. 7, 659 (2008); https://doi.org/10.1038/nmat2222
Y. Chen, J. Vela, H. Htoon, J. L. Casson, D. J. Werder, D. A. Bussian, V. I. Klimov, J. A. Hollingsworth, J. Am. Chem. Soc. 130, 5026 (2008). https://doi.org/10.1021/ja711379k
More about the authors
Fernando Stefani is a group leader in the department of physics at the Ludwig Maximilian University of Munich and the Center for NanoScience in Munich, Germany. Jacob Hoogenboom is an assistant professor of applied physics at Delft University of Technology in the Netherlands. Eli Barkai is a professor of physics at Barllan University in Ramat Gan, Israel, and a visiting scientist at the Massachusetts Institute of Technology in Cambridge.
Fernando D. Stefani, 1 Ludwig Maximilian University of Munich and the Center for NanoScience in Munich, Germany .
Jacob P. Hoogenboom, 2 Delft University of Technology, Netherlands .
Eli Barkai, 3 Bar-Ilan University in Ramat Gan, Israel .





{kind=link}
{kind=link}